ЭКО, дважды выявлена трисомия 13 хромосомы
Трисомия 13 хромосомы: что это такое, какая причина возникновения хромосомного заболевания?
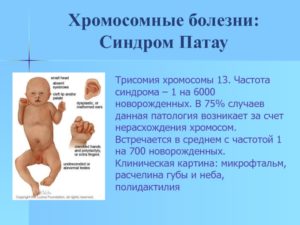
Трисомия 13 – тяжелая хромосомная патология, диагностируемая, в среднем, у одного ребенка из 7000. Такое заболевание, вызывающее внешние уродства и необратимые изменения в головном мозге, с одинаковой частотой встречается и у мальчиков, и у девочек. Согласно медицинской статистике, в 95% случаев дети, рожденные с аутосомной трисомией 13, гибнут в первые недели или месяцы жизни.
Каковы причины возникновения заболевания? По каким симптомам его распознают? Какие методы применяются при диагностике трисомии 13? Можно ли вылечить этот аутосомный синдром или предотвратить его развитие?
Трисомия по 13 хромосоме: что это такое?
Из школьного учебника биологии мы помним, что ядро живой клетки имеет своеобразный информационный «скелет», содержащий в себе генетический код. Эта структура называется кариотипом.
У человека она представлена в виде хромосомного набора из 23 диплоидных пар. 22 из них пронумерованы в порядке увеличения размера элементов.
Последняя, 23-яя, пара представляет собой комбинацию половых Х и Y хромосом, имеющую соответственное своей структуре наименование.В ряде генетических нарушений, носящих общее название «синдром трисомии», описанная выше система нарушается. Происходит это из-за образования лишнего, дополняющего уже сформированную гомологичную пару, хромосомного элемента. На сегодняшний день медиками всесторонне изучены случаи:
- Трисомии Х. Это специфическое заболевание затрагивает одноименную пару половых хромосом и, соответственно, наблюдается только у женщин. В зависимости от тяжести нарушения трисомия Х может выражаться через разные симптомы или не проявляться у пациентки вовсе.
- Синдрома Эдвардса. Так называют заболевание, возникающее из-за трисомии по 18 хромосоме. Пациенты с названным нарушением страдают от задержки умственного развития и лицевого дисморфизма.
- Трисомии 13. Это отклонение также носит название «синдром Патау» в честь доктора, первым объяснившего генетическую природу патологии. Рассматриваемое заболевание затрагивает 13 хромосомную пару. Из-за большого количества осложнений синдром Патау нуждается в более детальном (по сравнению с трисомией X и недугом, описанным Эдвардсом) рассмотрении.
Формы заболевания
При синдроме Патау дополнительная 13 хромосома может образоваться несколькими способами. Этим фактором обуславливается тип заболевания:
- Простой, когда лишняя хромосома свободно присутствует в организме, реализуя свой генетический потенциал.
- Форма робертсоновской транслокации, при которой дополнительный элемент образуется за счет слияния двух других; точнее – их генерирующих рРНК-плеч. При этом многократное повторение ошибок в кариотипе может не оказать никакого влияния на функциональность содержащих «неправильные» хромосомы клеток.
Обычно трисомия по 13 хромосоме является полной. Иными словами, нарушение затрагивает структурные ядра всех клеток организма. Реже встречается синдром Патау мозаичного типа.
Такая болезнь характеризуется мутациями некоторых клеток тела при нормальном строении остальных. В отдельных случаях трисомия рассматриваемого типа частична.
То есть у пациента обнаруживают не полноценную дополнительную хромосому, а лишь ее элементы в клеточном ядре.
Причины развития синдрома Патау
Согласно медицинским исследованиям, полное или частичное «утроение» 13-ой хромосомы происходит случайным образом еще в момент формирования родительских половых клеток. Факторов, однозначно способствующих появлению рассматриваемой генетической ошибки (включая наследственную предрасположенность), специалистам выявить не удалось.
Преклонный возраст матери, наличие у нее вредных привычек, а также иные негативные обстоятельства, такие как воздействие ионизирующего излучения или токсинов на плод, способны значительно повысить риск развития у ребенка синдрома Патау. Впрочем, верным утверждение останется в отношении любой трисомии, включая нарушение X типа.
Симптомы хромосомного заболевания
Тяжесть симптомов варьируется в зависимости от того, какая именно часть хромосомы была дублирована, но общая клиническая картина синдрома Патау остается идентичной для всех пациентов. Так, у младенцев болезнь может проявиться в виде:
- лицево-черепных деформаций, включая тригоноцефалию;
- измененного строения кистей и стоп (в редких случаях – с симметричной многопалостью);
- укороченной шеи;
- деформации и неправильного расположения глазниц и ушных раковин (иногда затрагивающей функциональность органов чувств);
- западающей переносицы;
- «заячьей губы»;
- крипторхизма (не опустившихся яичек) у мальчиков.
Методы диагностики
Визуальные симптомы синдрома Патау настолько характерны, что опытный неонатолог способен выявить болезнь уже в ходе беглого первичного осмотра младенца. Дополнительные исследования для подтверждения диагноза специалистам требуются редко.
С другой стороны, широкий комплекс существующих процедур позволяет обнаружить проблему еще на стадии вынашивания ребенка. При этом методы пренатальной диагностики синдрома Патау нисколько не отличаются от обследований, показанных для заблаговременного обнаружения утроения любых других парных элементов клеточного ядра (включая женские половые хромосомы Х).
Инвазивные исследования (амниоцентез, биопсия хориона)
Наиболее достоверным способом выявить синдром Патау на раннем сроке гестации считается лабораторный анализ биологического материала, взятого непосредственно из плодной оболочки зародыша. Подобные исследовательские методы называются инвазивными.
Наибольшее распространение в гинекологии получили следующие из них:
- Амниоцентез. В ходе процедуры в брюшную полость беременной вводится игла для забора биоматериала. Чтобы не допустить нанесения пациентке травм, доктор следит за происходящим по монитору аппарата УЗИ. Как правило, забор амниотической жидкости на анализ производят не позднее 14-18 недели гестации.
- Биопсия хориона. В данном случае лабораторному исследованию подвергают частички плацентарной ткани. Забор материала происходит по тому же сценарию, что и при амниоцентезе, но сама процедура из-за ее специфики считается более опасной для зародыша. Назначают хорионобиопсию лишь в крайних случаях и не позднее 10-12 недели вынашивания.
Комплексное исследование (скрининг)
По своей сути данный неинвазивный исследовательский метод является аналогом УЗИ. Однако, в отличие от упомянутой диагностической процедуры, скрининг предназначается для своевременного выявления конкретных генетических нарушений (утроения половой Х-хромосомы, синдромов Дауна, Эдвардса и т. д.).
Для исключения подозрений на болезнь Патау у ребенка будущие мамы вынуждены проходить обследование трижды, на разных сроках гестации:
- первичное – не ранее чем на 11 неделе;
- повторное – на 18-21 неделе;
- итоговое – не позднее 30-34 недели беременности.
Лечится ли заболевание, как можно предотвратить его возникновение?
Комплексное лечение, включающее множественные хирургические операции и медикаментозную терапию, способно лишь ненадолго продлить жизнь пациента, так и не сделав ее счастливой и полноценной.
Медиками зарегистрировано несколько случаев, когда больные синдромом Патау достигали хотя бы десятилетнего возраста, но и эти дети демонстрировали признаки серьезной умственной отсталости и страдали от проблем с функционированием внутренних органов.
Не приходится говорить и об успешной профилактике недуга. Непредсказуемость, с которой возникает патология, делает невозможным составления списка рекомендаций, соблюдение коих обезопасило бы будущее потомство от риска генетических нарушений.
Синдром Патау – что ожидает будущих родителей?

Врожденные заболевания, которые связаны с нарушением количества хромосом, хранящих наследственную информацию, встречаются примерно у 1 % новорожденных, при этом около 20 % спонтанных ранних выкидышей обусловлены аномальным набором хромосом у эмбрионов. Синдром Патау – одна из хромосомных патологий, несовместимых с нормальной жизнью.
Синдром Патау – что это за болезнь?
Синдром Патау характеризуется присутствием в клетках добавочной хромосомы номер тринадцать, т.е. вместо пары гомологичных хромосом данного типа присутствует три таких структуры. Аномалия также определяется термином «трисомия 13».
В норме набор хромосом в клетках человеческого организма (нормальный кариотип) представлен 46 элементами (23 пары), из которых две пары отвечают за половые признаки.
При исследовании кариотипа в клетках крови у любого человека могут быть выявлены изменения строения хромосом, не влияющих на его здоровье, но способных дать о себе знать у потомков.
Синдром Патау – тип наследования
При диагнозе «синдром Патау» кариотип записывается формулой такого вида: 47 XX (XY) 13+. При этом три копии тринадцатой хромосомы могут присутствовать во всех клетках тела, в других случаях дополнительная синтезированная хромосома встречается только в некоторых клетках.
Это случается вследствие ошибки при делении клеток в начале развития зародыша после соединения яйцеклетки и сперматозоида, возникающей под действием каких-либо внутренних или внешних воздействий.
Причем лишняя хромосома может исходить как от матери, так и от отца, не имеющих генетических отклонений.
Кроме этого, существуют случаи, когда дополнительная хромосома номер 13 может быть прикреплена к другой хромосоме в яйцеклетке или в сперматозоиде, что именуется транслокацией.
Это является единственной формой синдрома Патау, которая способна передаваться от кого-то из родителей.
Люди, являющиеся носителями измененного генетического материала и не имеющие признаков заболевания, могут передать его детям, которые родятся больными.
Риск трисомии 13
Синдром Патау у плода зачастую является горькой случайностью, от которой никто не застрахован.
В последнее время многим парам рекомендуют провести кариотипирование перед планированием зачатия, даже если не установлен высокий риск синдрома Патау или прочих хромосомных аномалий.
Эта методика изучает набор хромосом женщины и мужчины, выявляет различные отклонения. Как минимум, благодаря изучению генома родителей возможно спрогнозировать, есть ли вероятность наследственной формы патологии.
Как и многие другие хромосомные нарушения, рассматриваемая болезнь в большинстве случаев встречается у деток, зачатых женщинами старше 35-45 лет.
Поэтому на ранних сроках беременности назначается, если есть высокий риск синдрома Патау, амниоцентез – исследование клеток плода на наличие генетических дефектов.
Такой анализ осуществляется посредством пункционного прокола полости матки и забора околоплодных вод с наличием слущенных клеток плода.
Синдром Патау – частота встречаемости
Кариотип, свойственный синдрому Патау, регистрируется примерно один раз на каждые 7-14 тысяч новорожденных, появившихся на свет живыми. Встречаемость у мальчиков и у девочек одинаковая.
Кроме того, беременности с таким отклонением у плода относятся к группе высокого риска выкидыша или мертворождения.
В 75 % случаев родители малышей с данным диагнозом не имеют хромосомных отклонений, остальные эпизоды сопряжены с наследственным фактором – из-за передачи транслокализованой хромосомы номер 13 от одного из родителей.
Синдром Патау – причины возникновения
Ненаследственные формы заболевания пока не имеют четких причин возникновения, т.к. изучение провоцирующих факторов требует сложнейших исследований.
На сегодняшнее время даже не установлено точно, в какой момент происходит сбой – при формировании половых клеток или же при образовании зиготы.
Считается, что в дальнейшем дополнительная хромосома затрудняет чтение генома клетками, что становится препятствием для нормального формирования тканей, благополучного и своевременного завершения их роста и развития.Ученые предполагают, что синдром Патау причины может иметь связанные с такими факторами:
- воздействие ионизирующего излучения;
- токсическое воздействие;
- инфекционные заболевания;
- неблагоприятная экологическая обстановка;
- вредные привычки матери.
Синдром Патау – признаки
В некоторых случаях при поражении не всего количества клеток организма наблюдаются не слишком выраженные и не стремительно развивающиеся аномалии, но зачастую отклонения существенные. При этом, помимо внешних дефектов, которые можно увидеть на фото, синдром Патау характеризуется множеством пороков развития внутренних органов. Большинство отклонений имеют необратимый характер.
Перечислим, какие имеет синдром Патау симптомы:
1. Связанные с нервной системой:
- уменьшенный объем головы;
- нарушение формирования мозговых полушарий;
- отклонение психического развития;
- нарушение моторного развития;
- спинномозговой дефект;
- отсутствие органов зрения;
- катаракта;
- гипоплазия зрительного нерва;
- микрофтальмия;
- отслоение сетчатки.
2. Костно-мышечные, кожные отклонения:
- укороченная шея;
- искаженная форма ушных раковин;
- деформация стоп;
- пупочная грыжа;
- отсутствие участков кожи, волос;
- наличие лишних пальцев на руках и ногах;
- флексорное положение кистей;
- запавшая переносица;
- волчья пасть;
- заячья губа.
3. Урогенитальные симптомы:
- кисты в корковом слое почек;
- раздвоение мочеточников;
- аномалии гениталий.
4. Прочие аномалии:
- дефекты межсердечных перегородок;
- транспозиция магистральных сосудов;
- нарушения секреции гормонов;
- смещение поджелудочной железы;
- незавершенный поворот кишечника;
- добавочная селезенка и т.д.
Синдром Патау – методы диагностики
После рождения ребенка диагностика синдрома Патау у него не представляет никаких сложностей посредством визуального осмотра.
Для подтверждения диагноза производится анализ крови с целью обнаружить генотип синдрома Патау, ультразвуковое исследование.
Генетический анализ проводится и в случаях смерти младенца, что позволяет выяснить форму болезни, понять, не является ли она наследственной (важно при дальнейшем планировании детей).
Синдром Патау – анализы
При этом гораздо важнее своевременно установить отклонение на раннем этапе беременности, что возможно сделать примерно в конце первого триместра. До рождения трисомия по 13 хромосоме может быть выявлена путем исследования клеток из амниотической жидкости (околоплодных вод), полученных при проведении амниоцентеза или из клеток, полученных при биопсии хориона.
Пренатальный анализ может проводиться, когда родители входят в группу риска по развитию наследственной разновидности патологии и в рамках скрининга наследственной информации вынашиваемых плодов у беременных. На разных сроках применяются такие способы забора материала с целью проведения анализа методом количественной флуоресцентной полимеразной цепной реакции:
- с 8-12 неделю – взятие небольшого количества хориона (зародышевой оболочки);
- с 14-18 неделю – отбор околоплодных вод, содержащих клетки плода, через переднюю стенку брюшины;
- после 20 недели – пункция кровеносного сосуда пуповины.
Синдром Патау на УЗИ
Начиная с двенадцатой недели беременности, пороки развития у плода могут быть обнаружены посредством ультразвуковой диагностики. Для синдрома Патау характерно наличие следующих признаков:
- многоводие у беременной;
- маленькая асимметричная голова у плода;
- лишние пальцы на конечностях;
- утолщение воротниковой зоны;
- учащение частоты сердечных сокращений и др.
Лечение синдрома Патау
Как бы прискорбно это ни звучало, но дети с синдромом Патау являются неизлечимо больными, т.к. исправить хромосомные нарушения нельзя. Синдром Патау означает глубокую степень идиотии, полную инвалидность.
Родители, которые приняли решение родить малыша с такими отклонениями, должны настроиться на то, что ему потребуется постоянный уход и лечение.
Могут быть проведены оперативные вмешательства и медикаментозная терапия для корректировки функционирования жизненно важных систем и органов, пластические операции, профилактика инфекций и воспалений.
Синдром Патау – прогноз
Для детей, у которых диагностирован синдром Патау, продолжительность жизни в большинстве случаев не превышает одного года. Зачастую такие малыши обречены на летальный исход в первые недели или месяцы после появления на свет.
Менее 15 % деток доживают до пятилетнего возраста, а в развитых странах с высоким уровнем системы здравоохранения около 2 % пациентов доживают до десяти лет.
При этом даже те больные, у которых отсутствуют масштабные повреждения органов, не способны социально адаптироваться и самостоятельно заботиться о себе.
| Адреногенитальный синдром – все особенности патологии Адреногенитальный синдром – серьезное генетическое заболевание с опасными последствиями. Прочитайте о механизмах и причинах развития данной патологии, ее формах и клинических проявлениях. Узнайте, как своевременно выявить болезнь и правильно ее лечить. | Синдром Шерешевского-Тернера – каковы шансы на нормальную жизнь? В данной статье вы узнаете, что собой представляет Синдром Шерешевского-Тернера, как часто он встречается, какие причины его возникновения, симптомы и признаки, способы диагностики и лечения, а также последствия заболевания и прогнозы. |
| Сахарный диабет у детей прогрессирует намного быстрее, чем у взрослых и может привести к опасным осложнениям. Прочитайте, почему малыши страдают от этого эндокринного заболевания, научитесь распознавать его ранние симптомы и эффективно лечить. | Когда ребенку ставится диагноз «фенилкетонурия», что это за заболевание, родители зачастую узнают уже в первый месяц жизни крохи, после проведения неонатального скрининга. Раннее выявление патологии дает высокие шансы на ее успешное преодоление. |
Трисомия 13. Симптомы, диагностика, лечение

Другие названия Трисомии 13
Трисомия 13 является редким хромосомным расстройством, при котором вся или только часть хромосомы 13 проявляется в трех экземплярах (трисомия), а не в двух, в клетках организма.
У некоторых больных лиц, только некоторый процент клеток может содержать дополнительную 13-ю хромосому или ее часть (мозаицизм), в то время как другие клетки будут содержать нормальную хромосомную пару.
У лиц с трисомией 13, тяжесть симптомов и проявлений может сильно зависеть от конкретного местоположения дублированной (трисомной) части хромосомы, а также от процента клеток, содержащих эти хромосомные аномалии.
Тем не менее, у многих детей, такие аномалии могут включать в себя задержки в развитии, глубокую умственную отсталость, необычно маленькие глаза (микрофтальмия), заячью губу, расщелину в нёбе, неопущение яичек (крипторхизм) и дополнительные пальцы на руках и ногах (полидактилия).
Дополнительные аномалии головы и лица (черепно-лицевая область) также могут присутствовать, например, относительно небольшая голова (микроцефалия), широкий и плоский нос, широко расставленные глаза (гипертелоризм), вертикальные складки кожи в углах глаз, дефекты кожи на голове и аномальные, низко посаженные уши.
Дети могут также иметь неполное развитие определенных областей мозга (например, передний мозг), почек и структурные аномалии сердца. Например, характерные пороки сердца могут включать в себя отверстие в перегородке, разделяющей верхние и нижние камеры сердца или сохранение отверстия между двумя основными сосудами — аорта и легочная артерия.
Многие дети с трисомией 13 перестают расти и набирать вес. Также они имеют серьезные трудности в кормление, снижение мышечного тонуса (гипотония) и эпизоды спонтанного апноэ. Опасные для жизни осложнения также могут развиться.
Трисомия 13. Эпидемиология
Трисомию 13 иногда называют синдром Патау, который был назван так в честь доктора Патау, определившего трисомное происхождение этого синдрома в 1960 году. Этот синдром развивается у лиц женского пола несколько чаще, чем у лиц мужского пола и развивается он примерно у одного ребенка на 5000-12000 живорожденных.
Опыт показывает, что примерно один процент всех признанных выкидышей происходит в связи с трисомией 13. Кроме того, как отмечалось выше, частота трисомии 13 увеличивается с возрастом матери.
Исследователи также предложили возможную связь между преэклампсией и трисомией 13. Преэклампсия является ненормальным состоянием беременности, которое характеризуется быстрым развитием высокого кровяного давления (гипертония), высвобождением аномального количества белка в мочу (протеинурия), и / или чрезмерной задержкой жидкости (отек).
По мнению исследователей, число случаев преэклампсии, кажется, значительно выше у женщин у которых развивается плод с трисомией 13. Кроме того, частота развития преэклампсии при этой трисомии значительно выше, чем при некоторых других хромосомных аномалиях (например, трисомия 18, трисомия 21 [Синдром Дауна]).
Исследователи предполагают возможность того, что именно ген или гены на хромосоме 13, могут влиять на развитие преэклампсии.
Трисомия 13. Причины
У лиц с трисомией 13, вся или относительно большая область хромосомы 13, присутствует в качестве третьей копии (трисомия), а не в двух экземплярах, как это должно быть. Примерно в пяти процентах случаев, такая аномалия будет содержать только в определенном проценте клеток (мозаицизм), в то время как в других клетках будет нормальный набор хромосом.
Тяжесть и спектр симптомов и проявлений может зависеть от длины дублированной части хромосомы. Кроме того, лица с мозаицизмом, как правило, имеют менее тяжелые симптомы. Однако, в таких случаях, проявления болезни могут быть очень разнообразными, начиная от почти незаметных и легких и заканчивая тяжелыми пороками.
У большинства людей с трисомией 13, дублирование хромосомы 13 или ее части, вызывается спонтанными (De Novo) ошибками во время деления половых клеток в одном из родителей (например, во время нерасхождения при мейозе). Опыт показывает, что риск развития таких ошибок может увеличить у уже возрастных лиц.
Примерно 20 процентов лиц, развивают трисомию 13, в результате транслокации с участием хромосомы 13 и другой хромосомы.Исследователи также предполагают, что определенные признаки и проявления, связанные с трисомией 13 могут быть следствием избыточной экспрессии важных генов в хромосоме 13.
Например, можно выделить ген, который регулирует выработку фермента, известного как эстераза D, этот очень важный ген расположен на длинном плече хромосомы 13 (13q14.11).
Повышенные уровни эстераз D были обнаружены в тканях почек у некоторых младенцев.
Трисомия 13. Фото
Циклопия у ребенка с трисомией 13
Новорожденный мальчик с полной трисомией 13 (синдром Патау). Этот ребенок имеет расщелину нёба, паховую грыжу и полидактилию на левой руке.
Ребенок с трисомией 13
Крупная двусторонняя заячья губа у ребенка с трисомией 13.
Трисомия 13. Симптомы и проявления
Сопутствующие симптомы и проявления могут сильно отличаться от случая к случаю. Тем не менее, трисомия 13 часто характеризуется черепными, неврологическими, сердечными и / или другими дефектами.
Дети, с этим синдромом, как правило, необычайно малы и имеют проблемы с кормлением.
Эти дети могут иметь различные краниофациальные пороки, например, аномально маленькая голова (микроцефалия) и наклонный лоб, необычная ширина родничков на передней и задней части черепа, расщелина нёба, небольшие челюсти, язвы на коже в верхней части головы и / или низко посаженные и деформированные уши.
Другие проявления могут включать в себя короткую шею, складки кожи на задней части шеи и / или наличие доброкачественных родинок или поражений, состоящих из кластеров аномальных кровеносных сосудов (капиллярные гемангиомы), которые наиболее часто проявляются в центре лба.
Кроме того, у многих детей могут быть аномалии глаз, к ним можно отнести необычно маленькие глаза (микрофтальмия), частичное отсутствие глазной ткани, аномальное развитие сетчатки (дисплазия), вертикальные складки кожи над внутренними углами глаз и / или другие глазные дефекты. Кроме того, брови могут быть редкими или они вообще могут отсутствовать.
Трисомия 13 также часто характеризуется таким состоянием, при котором передний мозг не в состоянии должным образом разделиться во время эмбрионального развития.
У лиц с этой проблемой могут развиться другие, различные сопутствующие дефекты средней линии лица, в том числе, тесно посаженные глаза, аномалии носа и / или другие проявления.
Еще, очень редко, у некоторых детей может развиться циклопия, которая характеризуется слиянием глазных впадин (орбит) в единую полость, в которой будет находиться или один или оба глаза.
Младенцы также могут иметь дополнительные аномалии центральной нервной системы (т.е., головного и спинного мозга). Дети могут испытывать временные эпизоды апноэ или внезапные и неконтролируемые судороги.Многие младенцы имеют глухость и глубокую умственную отсталость.
Кроме того, в некоторых случаях, дополнительные проявления могут включать в себя ненормальный тон мышц, агенезию мозолистого тела, неразвитость мозжечка (мозжечковая гипоплазия), гидроцефалию и / или миеломенингоцеле.
Около 80% детей с трисомией 13 также имеют врожденные пороки сердца, такие как дефекты перегородок или открытый артериальный проток. Кроме того, у некоторых детей сердце может быть расположено в правой стороне груди (декстрокардия).
Почечные дефекты также могут развиться. Они могут включать множественные кисты в почках, подковообразность почек и / или отек почек из-за закупорки или сужения мочеточников (гидронефроз). Некоторые аномалии половых органов также связаны с трисомией 13, в том числе крипторхизм, аномально сформированные мошонки и слаборазвитые яичники, матки.
Младенцы с трисомией 13 также часто имеют определенные отклонения в руках и ногах. Они могут включать в себя аномальное количество пальцев рук и / или ног (полидактилия), возможно срощение некоторых пальцев и необычно округлые ногти.
В некоторых случаях, другие нарушения также могут присутствовать. Такие признаки могут включать в себя тонкие ребра, таз, слаборазвитость мышц, грыжи, аномальное развитие поджелудочной железы и / или другие аномалии.
Трисомия 13. Похожие расстройства
Симптомы следующих расстройств могут быть аналогичны трисомии 13. Сравнения могут быть полезными для дифференциальной диагностики:
Псевдо-трисомия 13 — это редкое заболевание характеризуется развитием аномалий в средней лицевой линии, дополнительными пальцами рук и / или ног (полидактилия), пороками сердца. Недоразвитие мозолистого тела, гидроцефалия и другие особенности также могут присутствовать у лиц с этим расстройством.
Существует также целый ряд других расстройств, в том числе других хромосомных синдромов, которые могут быть охарактеризованы симптомами и проявлениями, которые развиваются у лиц с трисомией 13.
Трисомия 13. Диагностика
В некоторых случаях, диагноз трисомии 13 может быть поставлен еще до рождения (пренатально). После проведения специализированных тестов, таких как УЗИ плода, амниоцентез и / или биопсия хориона.
Например, во время проведения УЗИ можно обнаружить некоторые проявления, которые могут навести на мысль о наличии у плода трисомии 13, эти аномалии включают полидактилию и замедление роста.
При проведении хромосомных исследований, можно выявить наличие дополнительной хромосомы 13.Диагноз трисомии 13 также может быть подтвержден после рождения, с помощью тщательного клинического обследования, выявления характерных физических проявлений и хромосомного анализа. Во время проведения лабороторных тестов можно также выявить необычное сохранение эмбрионального и / или фетального гемоглобина в крови у новорожденных и грудных детей с трисомией 13.
Трисомия 13. Лечение
Лечение трисомии 13 направлено только в сторону контроля конкретных симптомов, которые проявляются в каждой особи по разному. Такой план лечения может потребовать скоординированных усилий целой команды медицинских специалистов.
В некоторых случаях, рекомендуемое лечение может включать хирургическую коррекцию некоторых нарушений, связанных с расстройством. Хирургические процедуры будут зависеть от природы и тяжести анатомических нарушений, связанных с ними симптомами и от других факторов.
Физиотерапия, генетическое консультирование и другие подходы будут полезными для семей, имеющих детей с трисомией 13. Все типы лечения этого расстройства будут только симптоматическими и поддерживающими.
Синдром Патау — Трисомия 13

Блог / Генетика и болезни / Синдром Патау — Трисомия 13
Синдром Патау – это хромосомное заболевание, обусловленное наличием лишней хромосомы 13 пары.
Данная патология была открыта в 1657 году ученым Эразмусом Бартолином, однако хромосомная причина была доказана только в 1960 году доктором Клаусом Патау.
Синдром Патау относится к редким патологиям (1:7000-1:10000 случаев), с одинаковой частотой встречается у обоих полов.
Наследственная этиология заболевания не доказана, частота возникновения увеличивается с возрастом родителей.
Узнать с точностью 99% о риске синдрома Патау и других хромосомных аномалий, а также пол плода, можно с 9 недель беременности, всего лишь сдав кровь из вены. Подробнее о НИПТ-тесте.
Признаки Синдрома Патау
- низкая масса тела при доношенной беременности (менее 2500 грамм);
- неправильное строение черепа (небольшие размеры, сужение лба, расширение затылочной области);Выраженные отклонения физического и умственного развития;
- пороки развития структур головного мозга;
- пороки развития глаз (отсутствие глазных яблок, маленькие глаза, катаракта, отслойка сетчатки и др.);
- заячья губа;
- волчья пасть;
- деформированная форма ушей;
- пороки развития кисти (лишние пальцы, неправильное формирование большого пальца);
- локальное отсутствие кожи, волос;
- деформированные стопы, лишние пальцы на ногах;
- множественные пороки мочевыводящей, сердечно-сосудистой, пищеварительной и половой систем.
Прогноз
Прогноз при синдроме Патау неблагоприятный. Из-за наличия множественных пороков развития большинство детей погибают в первые месяцы после рождения (95 % пациентов). Дети, оставшиеся в живых, имеют резкие нарушения психомоторного развития (вплоть до глубокой идиотии).
Пренатальная диагностика
1) Инвазивные исследования (амниоцентез, биопсия хориона) в основном назначают тем женщинам, у которых наблюдается повышенный риск того, что родится малыша с синдромом Эдвардса, например, пациенткам, чей возраст превышает 35 лет или с плохими результатами неинвазивных тестов: УЗИ и анализов. Инвазивные методы диагностики являются высокоточными, однако, учитывая риск осложнений, не подходят для массового проведения всем беременным, а проводятся только по особым показаниям.
2) Неинвазивные технологии, так называемые скрининги. Скрининг – комплексное исследование беременных женщин на наличие у плода хромосомных аномалий.
Выделено несколько признаков, указывающих на высокий риск наличия заболевания, которые может выявить УЗИ плода (отсутствие носовой кости, увеличенная толщина воротникового пространства, недостаточная длина бедренных и плечевых костей и другие особенности). В комплексе с УЗИ идёт биохимический анализ крови матери на такие гормоны как свободный бета-ХГЧ и PAPP-A.
Полученные данные по биохимическим маркерам анализируют в совокупности с результатами ультразвукового исследования, а результат всего скрининга представляет собой расчет риска наличия хромосомной аномалии у плода.
Однако при использовании стандартных тестов на синдром Эдвардса, лишь у 3% женщин, направленных на инвазивную диагностику действительно подтверждается наличие заболевания. В то же время не исключены и ложно-отрицательные результаты, когда скрининг показывает низкий риск, а ребенок рождается с хромосомной патологией.
Неинвазивный метод исследования (НИПТ-тест)
- точность 99%, что намного точнее классической диагностики (УЗИ и биохимический скрининг)
- совершенно безопасен, в отличие от инвазивных методик — для забора материала на анализ необходимо просто взять кровь из вены беременной женщины.
- на ранних сроках: анализ можно проводить уже на 9-й неделе беременности.
Узнать подробнее
Дата: 2016-07-03
Ваш голос учтен!
Получите бесплатную консультацию по анализам ДНК
Самые частые патологии плода во время беременности. Обзор синдромов
Пренатальная диагностика — это комплекс генетических исследований во время беременности, проводящийся с целью обнаружения патологии у плода на стадии внутриутробного развития.
Данный вид исследований позволяет обнаружить более 99 % плодов с синдромом Дауна (или трисомия по 21 хромосоме), синдромом Эдвардса (трисомия по 18 хромосоме),синдромом Патау (трисомия по 13 хромосоме). Хромосомные аномалии имеют место в 1 из 150 случаев деторождения.
В случае наличия у плода болезни родители при помощи врача-генетика тщательно взвешивают возможности современной медицины и свои собственные в плане реабилитации ребёнка.
В результате семья принимает решение о судьбе данного ребёнка и решает вопрос о продолжении вынашивания
Пренатальная диагностика подразделяется на инвазивную и неинвазивную
- Инвазивные методы диагностики – такие как биопсия хориона, амниоцентез (забор околоплодных вод), кордоцентез (забор крови из пуповины).
- Неинвазивные методы диагностики более щадящие. К ним относят неинвазивный пренатальный тест на частые хромосомные аномалии, которые совместимы с жизнью, но такие хромосомные аномалии как синдромы Дауна, Эдвардса и Патау сопровождаются множественными пороками развития и не поддаются излечению. НИПТ надежнее биохимического скрининга, так как относится к прямым методам исследования плодной ДНК. При этом, у будущей мамы нет риска напрасной тревоги при беременности при попадании в группу риска по хромосомным аномалиям, нет риска спонтанного прерывания беременности. НИПТ одобрен во многих странах и включен в систему добровольного медицинского страхования (такие развитые страны как Швейцария, США, Канада, Нидерланды, Германия).
Какие же основные синдромы выявляет НИПТ
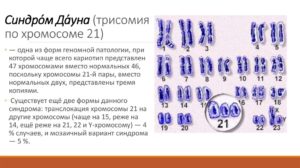
- Синдром Дауна (трисомия 21)
Частота встречаемости — 1:600 родов. Причем, риск трисомии 21 у плода увеличивается с возрастом женщины.
Одна из форм геномной патологии, при которой чаще всего кариотип представлен 47 хромосомами вместо нормальных 46, поскольку хромосомы 21-й пары, вместо нормальных двух, представлены тремя копиями.
Существует ещё две формы данного синдрома:транслокация хромосомы 21 на другие хромосомы (чаще на 15, реже на 14, ещё реже на 21, 22 и Y-хромосому) — 4% случаев, и мозаичный вариант синдрома — 5%.Характерные особенности, сопутствующие синдрому Дауна: «плоское лицо», аномальное укорочение черепа, кожная складка на шее у новорожденных, короткие конечности, открытый рот и другие признаки, которые может точно диагностировать врач-генетик.
- Синдром Эдвардса (трисомия 18)
Хромосомное заболевание характеризуется комплексом множественных пороков развития и трисомией 18 хромосомы. Описан в 1960 году Джоном Эдвардсом. Популяционная частота по миру 1:5000 на 2016 г.
Дети с трисомией 18 чаще рождаются у пожилых матерей, взаимосвязь с возрастом матери менее выражена, чем в случаях трисомии 21 хромосомы.
Основные признаки синдрома Эдвардса: малый вес, аномалии форм черепа, пороки сердца, дефекты межжелудочковой перегородки, умственная отсталость.
- Синдром Патау (трисомия 13)
Частота – 1:16000 родов. Характерны нарушения развития нервной системы, глаз, выраженная деформация лица, пороки развития сердца и других внутренних органов. Характерна глубокая задержка умственного развития. 90% детей умирают в возрасте до 1 года.
Гоносомные анеуплоидии:
- Синдром Шерешевского-Тернера
Если в клетке присутствует только одна хромосома вместо двух, это называется моносомией. Частота встречаемости – 1:2500.
Возникает в случае отсутствия второй половой хромосомы, встречается только у девочек. Единственная моносомия, при которой возможно рождение живого ребенка.
Хромосомная болезнь, сопровождающаяся характерными аномалиями физического развития, низкорослостью и половым инфантилизмом.
Частота – 1:500. Когда в клетках с мужским кариотипом присутствует дополнительная Х-хромосома. Встречается только у мальчиков. Это заболевание, при котором особи мужского пола имеют дополнительную Х-хромосому.
Обычно женщины имеют пару ХХ хромосом, а мужчины пару ХY хромосом, однако при этом заболевании мужчины имеют по крайней мере две Х-хромосомы и хотя бы одну Y хромосому.
Синдром Клайнфельтера является не только самой частой формой мужского гипогонадизма, бесплодия, эректильной дисфункции, но и одной из наиболее распространённых эндокринных патологий, занимая третье место после сахарного диабета и заболеваний щитовидной железы.
- Синдром XYY (синдром Джейкобса)
Частота – 1:1000. В клетках присутствует дополнительная Y-хромосома. Встречается только у мальчиков. Носитель синдрома имеет дополнительную Y-хромосому, общий хромосомный набор составляет 44 аутосомы и три половые хромосомы. Наличие второй Y-хромосомы в большинстве случаев не ведёт к каким-либо физическим отклонениям.
В то же время, многие мужчины с XYY-синдромом имеют одну или несколько особенностей. При рождении они имеют нормальный рост, но часто быстрее растут в детстве. В среднем, во взрослом состоянии носитель выше, чем 75% мужчин того же возраста. Некоторые мужчины с синдромом XYY имеют небольшие нарушения координации движений, в результате чего могут казаться неуклюжими.
Фертильность чаще всего не нарушена, обычно такие мужчины гетеросексуальны и имеют нормальную сексуальную функцию. Тем не менее, описаны случаи существенного снижения фертильности, вплоть до бесплодия. У небольшого числа носителей также повышен уровень половых гормонов, связанных со сперматогенезом, что может вести к бесплодию ввиду нарушения образования спермы.
Неизвестно, насколько высоко число случаев бесплодия у мужчин с XYY-синдромом. IQ находится в пределах нормы, но часто несколько ниже, чем у родных братьев и сестёр. Примерно половина носителей имеет проблемы с обучением, в частности, могут быть нарушения и чтения.
Может быть повышен риск поведенческих проблем, таких как синдром гиперактивности, мужчины с XYY-синдромом часто импульсивны и эмоционально незрелы.
- Синдром тройной Х-хромосомы
Частота – 1:1000. В клетках присутствует 3 копии Х-хромосомы. Встречается только у девочек.
В большинстве случаев носители дополнительной X-хромосомы — женщины без заметных признаков патологии, поэтому при медицинских исследованиях 90% трисомиков по X-хромосоме остаются не выявленными.
Развитие может протекать с некоторыми нарушениями, могут возникнуть проблемы с координацией, моторикой и развитием речи. В некоторых случаях отмечен меньший размер головы (без заметного снижения умственных способностей).
Трисомия по X-хромосоме не приводит к значительным нарушениям фертильности, в большинстве случаев может проявляться только в более ранней менструации.Синдром Патау (трисомия 13): первые симптомы у новорожденных, лечение и профилактика
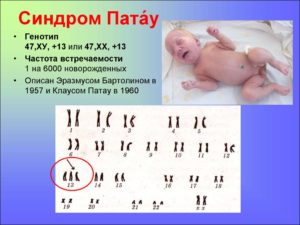
Синдром Патау (трисомия 13) — хромосомное заболевание человека, которое характеризуется наличием в клетках дополнительной хромосомы 13.
Синдром Патау – генетическая аномалия, для которой характерно наличие у ребёнка многочисленных пороков строения и функционирования органов и систем, головного мозга и опорно-двигательного аппарата.
Обуславливается возникновение такого расстройства наличием запасной тринадцатой хромосомы.
Для недуга характерны дефекты центральной нервной и сердечно-сосудистой систем, органов зрения и слуха, мышечной системы (наличие расщелины губы), патологии строения органов половой системы.
Причины
Установлено, что причиной синдрома Патау, в большинстве случаев, является утроение 13 хромосомы, то есть в каждой клетке имеется не две (норма), а три копии тринадцатой хромосомы. В очень редких случаях только часть клеток организма имеет дополнительную копию. Это так называемый мозаичный синдром Патау.
Еще одна причина заболевания – транслокация (перестройка) хромосом, когда часть 13 хромосомы до или непосредственно в момент зачатия привязывается к другой негомологичной хромосоме.
В результате такой перестройки у больных наряду с двумя копиями 13 хромосомы появляется дополнительный материал из нее, который подключен к другой хромосоме.
Возникает частичная трисомия 13 хромосомы, при которой физические признаки синдрома несколько отличаются от типичной клинической картины.
Как правило, синдром Патау не наследуется, а возникает случайно в процессе формирования сперматозоидов и яйцеклеток. Если при делении клеток возникает ошибка, которая называется нерасхождением, это приводит к появлению половых клеток с неправильным числом хромосом.
Когда подобные атипичные сперматозоиды и яйцеклетки вовлекаются в генетическую структуру ребенка, он получает дополнительную 13 хромосому во всех клетках организма.
Мозаицизм данного синдрома также не наследуется и возникает как случайный сбой при делении клеток на первоначальном этапе развития зародыша.
Симптомы
Далее опишем симптомы болезни Патау, которые можно заметить как во время беременности, так и после рождения малыша.
При беременности Синдром Патау имеет следующие признаки во время беременности:
- Время пребывания плода в утробе сокращается до 38 недель.
- Возникает многоводие (количество околоплодных вод превышает норму).
- В процессе вынашивания может быть выкидыш. Ребёнок рождается мёртвым.
У новорожденных
Далее перечислим характерные симптомы синдрома:
- Ребёнок рождается недоразвитым, что проявляется как умственными, так и физическими отклонениями.
- Врождённые пороки (мутации и уродства).
- Нарушения в развитии нервной системы.
- Деформация рук.
- Деформация черепа и уродства лица.
- Западание переносицы.
- Помутнение роговицы глаз.
- Расщелина в верхнем нёбе и губе.
- Формирование лишней селезёнки.
- Деформации половых органов.
Это не полный список всех возможных последствий, однако, даже перечисленные выше опасности говорят о том, что ребёнок может родиться с такими уродствами, что не будет похож на человека. В худшем случае младенец из-за мутаций может умереть сразу после родов.
Диагностика патологии
Для диагностики этого недуга проводится исследование кариотипа малыша сразу же после рождения. Существуют методики обнаружения этого расстройства у плода, находящегося внутри утробы матери. Делают это при помощи УЗИ — специфические проявления наблюдаются начиная с двенадцатой недели беременности.
Заболевание встречается в равной степени как у мальчиков, так и у девочек. По степени распространённости недуг уступает только синдрому Дауна — с таким расстройством рождается один малыш на десять тысяч. Специальных методик лечения и устранения этого синдрома не существует.
Единственное, что могут сделать врачи, провести коррекцию врождённых патологий.
Выявить наличие синдром у плода можно на УЗИ, начиная с 20 недель беременности. При выявлении различных нарушений развития плода (неправильный череп, слишком низкая масса, расщелина верхней губы, отсутствие волос и т. д.). После рождения диагноз подтверждается врачами исходя из осмотра ребенка и клинической картины.
Методы диагностики:
- УЗИ;
- исследование кариотипа;
- биопсия ворсинок хориона;
- амниоцентез.
Лечение
Возможности медицинской помощи детям с синдромом Патау ограничены и сводятся, главным образом, к организации хорошего ухода, полноценного питания, профилактике инфекций, общеукрепляющей и симптоматической терапии. Хирургическая помощь может потребоваться для устранения врожденных пороков сердца, расщелин лица и др.
Дети с синдромом Патау находятся под наблюдением педиатра, детского генетика, детского невролога, детского кардиолога, детского офтальмолога, детского травматолога-ортопеда, детского отоларинголога, детского гастроэнтеролога, детского уролога и других специалистов.
Профилактика
По причине неточной этиологии возникновения синдрома, специфической профилактики не существует, но есть несколько полезных рекомендаций, позволяющих избежать рождения малыша с расстройствами кариотипа: по возможности сменить район, город или даже страну проживания; избегать каких-либо контактов с химическими веществами; не иметь половых контактов с близкими родственниками. В случаях, когда это всё-таки произошло, не рожать ребёнка; не беременеть после сорока пяти лет. Если будущие родители не знают, являются ли они носителями какого-либо генетического расстройства, лучшим решением будет прохождение консультации у специалиста в данной области ещё до зачатия ребёнка, на этапе планирования.
(1 5,00 из 5)
Загрузка…