Трисомия по 22 хромосоме у эмбриона
Кольцевая 22 хромосома. Симптомы, диагностика, лечение

Кольцевая 22 хромосома – редкое расстройство, которое характеризуется аномалиями в 22 хромосоме. Сопутствующие симптомы и проявления могут быть очень разнообразными, от случая к случаю.
Тем не менее, это расстройство, как правило, связано с развитием умственной отсталости от умеренной до тяжелой степени. Некоторые лица могут также иметь относительно мягкие, неспецифические физические (т.е.
диспластические) проявления, в то время как другие могут иметь более отличительные, потенциально серьезные физические отклонения.
Согласно информации, указанной в медицинской литературе, общие для всех пациентов проявления включают уменьшенный мышечный тонус (гипотония), нарушение координации, необычная манера ходьбы, трудности в речевой функции и / или некоторые пороки развития черепа и лицевой (черепно-лицевой) области.
Такие черепно-лицевые аномалии могут включать в себя необычно маленькую голову (микроцефалия), аномальные складки кожи, которые могут охватывать внутренние углы глаз, необычно большие уши и / или другие пороки развития. Кольцевая 22 хромосома, как правило, вызывается спонтанными или «De Novo» ошибками в самом начале развития эмбриона, которые происходят случайным образом и пока по неизвестным причинам.
Кольцевая 22 хромосома. Эпидемиология
На основании наблюдаемых случаев, кольцевая 22 хромосома развивается у женщин чаще, чем у мужчин. В медицинской литературе зарегистрировано более 50 случаев.
Кольцевая 22 хромосома. Причины
Кольцевая 22 хромосома развивается из-за потери кусочков хромосомы на обоих концах 22 хромосомы, с последующим соединением этих концов.
У лиц с кольцевой 22 хромосомой, сопутствующие симптомы и проявления могут быть очень разнообразны, в зависимости от количества потерянного генетического материала в 22 хромосоме, стабильности кольцевой хромосомы во время последующих клеточных делений (т.е. митоза) и от других факторов.
Кольцевая 22 хромосома, как правило, развивается из-за спонтанных или «De Novo» ошибок в самом начале развития эмбриона. В таких случаях, родители больного ребенка, как правило, имеют нормальные хромосомы.
Тем ни менее, были зарегистрированы случаи, в которых кольцевые 22 хромосомы были унаследованы от родителей (семейная передачи).В некоторых случаях, только определенный процент клеток родителя может содержать кольцевые 22 хромосомы, в то время как другие клетки могут иметь нормальные хромосомы (такое событие известно как «мозаицизм»).
Кольцевая 22 хромосома. Симптомы и проявления
Кольцевая 22 хромосома, как правило, характеризуется развитием умственной отсталости, связанной с различными физическими проявлениями, которые могут варьироваться от относительно мягких и неспецифических до более отличительных и потенциально серьезных для здоровья. Отчеты показывают, что физическое развитие и рост находятся норме даже у наиболее пострадавших лиц.
Девочка с кольцевой 22 хромосомой
В дополнение к умственной отсталости, общие черты, связанные с кольцевой 22 хромосомой включают низкий мышечный тонус (гипотония), плохую координацию, неуклюжую и неустойчивую походку, речевые проблемы. Некоторые лица также могут иметь поведенческие отклонения, такие как заметно повышенная двигательная активность и аутизм.
Многие люди с кольцевой 22 хромосомой также развивают пороки черепа и пороки лицевой (черепно-лицевой) области.
Они обычно включают в себя аномально маленькую голову (микроцефалия), относительно длинное лицо, толстые, низкие брови, вертикальные складки кожи, которые могут охватывать внутренние уголки глаз и / или большие уши.
Некоторые лица также могут иметь дополнительные черепно-лицевые дефекты, такие как большой нос в форме картошки, толстые, полные губы, широко расставленные глаза и / или короткие, узкие складки век. Другие особенности могут включать птоз, высокое нёбо и / или высунутый язык.
Некоторые лица могут также иметь нарушения рук и ног. Такие нарушения могут включать в себя срощение определенных пальцев рук или ног, особенно это касается второго и третьего пальца, недоразвитие костей, ногтей, тонкие пальцы и/или необычно большие руки и ноги.
Мальчик с кольцевой 22 хромосомой
В редких случаях могут наблюдаться другие, более серьезные физические аномалии, такие как структурные пороки развития сердца (врожденные сердечные дефекты), дефекты почек, частичное или полное закрытие анального отверстия тонкой мембраной или ненормальное накопление лимфы в тканях и связанный с ним отек (лимфедема).
В некоторых случаях, некоторые из особенностей, связанные с кольцевой 22 хромосомой, могут напоминать те, которые развиваются у людей с моносомией 22 или с синдромом кошачьего глаза.
Кольцевая 22 хромосома. Похожие расстройства
Симптомы и проявления следующих расстройств могут быть аналогичны тем, которые развиваются у лиц с кольцевой 22 хромосомой. Сравнения могут быть полезными для дифференциальной диагностики:
Моносомия по 22 хромосоме. Это редкое расстройство характеризуется полным отсутствием одной 22-й хромосомы или ее части. В большинстве случаев, у пациентов отсутствует длинное плечо 22-й хромосомы, а у небольшого числа пациентов только часть в этом длином плече.
Нарушения могут быть переменными, всё зависит от конкретного места хромосомной делеции.
Тем не менее, многие пациенты могут иметь умственную отсталость, тяжелые задержки речи, дефицит роста, снижение мышечного тонуса (гипотония), и / или черепно-лицевые пороки развития – аномально маленькую голову (микроцефалия), большие уши, плоский носовой мост, широко разнесенные глаза (глазной гипертелоризм), вертикальные складки кожи и / или другие проявления. У некоторых пациентов также может быть отсутствие (агенезия) тимуса и паращитовидных желез. Тимус играет важную роль в развитии иммунной системы в период от внутриутробного развития и до периода полового созревания.Синдром Диджорджи. Этот синдром связан с делецией в локусе 22q11.2. Синдром характеризуется задержками развития, ускоренным ростом, речевыми задержками, мышечной слабостью (миотония) и мягкими структурными дефектами (дисморфизмы).
Синдром кошачьих глаз является редким хромосомным расстройством, при котором одни люди могут иметь легкие симптомы и проявления, в то время как другие могут иметь более серьезные пороки развития, особенно черепно-лицевой области, желудочно-кишечного тракта, сердца и / или почек.
Аномалии лицевой области могут включать в себя депрессию носа, косые складки век, широко расставленные глаза, односторонние или двусторонние колобомы, небольшие челюсти (микрогнатия) и / или ненормальные наросты кожи и мелкие углубления перед ушами.
Дополнительные физические аномалии могут включать структурные аномалии сердца, неразвитость (гипоплазия) и / или отсутствие (агенезия) почек, невысокий рост и / или другие особенности. Расстройство часто связано с умственной отсталостью.
Кольцевая 22 хромосома. Диагностика
В некоторых случаях диагноз кольцевой 22 хромосомы может быть поставлен еще до рождения (пренатально) при выполнении УЗИ, амниоцентеза, и / или биопсии хориона.
С помощью УЗИ можно выявить характерные проявления, которые будут свидетельствовать о хромосомных аномалиях.
После чего, врачи могут провести амниоцентез (сбор околоплодной жидкости) или сбор ворсинок хориона для последующего хромосомного анализа, во время которого будет определена кольцевая структура 22 хромосомы.Диагноз также может быть подтвержден после рождения, на основе тщательного клинического обследования, выявления характерных физических проявлений и на основе того же хромосомного анализа.
Кольцевая 22 хромосома. Лечение
Лечение этого расстройства направлено только на контроль и избавление от конкретных симптомов и проявлений. Такое лечение может потребовать скоординированных усилий команды медицинских работников, таких как педиатров, хирургов и других специалистов.
Для некоторых пациентов, лечение может включать в себя хирургическое вмешательство для исправления некоторых черепно-лицевых или других физических аномалий, потенциально связанных с этим расстройством. Типы и количество хирургических процедур, будут зависеть от тяжести анатомических нарушений, связанных с ними симптомов и других факторов.
Раннее вмешательство может быть важным для любого из таких пациентов. Для детей также будут полезны физическая терапия, логопедия и / или другие медицинские, социальные и / или профессиональные направления. Генетическое консультирование также будет полезным для людей с кольцевой 22 хромосомой и их семей.
Синдром делеции 22 хромосомы (синдром ДиДжорджи) у детей. Клинические рекомендации

- Аутоиммунные осложнения
- Велофарингеальная недостаточность
- Врожденный порок сердца
- Гипокальциемия
- Гипопаратиреоз
- Делеция 22 хромосомы
- Дефект Т-клеточного звена
- Задержка речевого и психомоторного развития
- Задержка физического развития
- Внутривенные иммуноглобулины
- Инфекционные осложнения
- Расщепление неба и верхней губы
- Синдром ДиДжорджи
АИГА – аутоиммунная гемолитическая анемия
АЛТ — аланинаминотрансфераза
АСТ — аспартатаминотрансфераза
ВВИГ — внутривенные иммуноглобулины
ВЗК – воспалительные заболевания кишечника
ВПС – врожденный порок сердца
ГКС — глюкокортикостероиды
ДНК — дезоксирибонуклеиновая кислота
ЖКТ — желудочно-кишечный тракт
ИТП – иммунная тромбоцитопения
КМ — костный мозг
КТ — компьютерная томография
ЛОР — ларингооторинолог
ЛПУ — лечебно-профилактическое учреждение
МЗ — Министерство здравоохранения
МКБ-10 — Международная классификация болезней 10-го пересмотра
МРТ —магнитно-резонансная томография
РКИ — рандомизированные контролируемые исследования
РНК — рибонуклеиновая кислота
РФ — Российская Федерация
Синдром del 22q11 – синдром делеции 22 хромосомы=синдром ДиДжорджи
СДД — синдром ДиДжорджи
ТГСК – трансплантация гемопоэтических стволовых клеток
УЗИ – ультразвуковое исследование
ЭКГ – электрокардиограмма
ЭхоКГ -эхокардиография
ЮРА – ювенильный ревматоидный артрит
del 22q11.2 – делеция длинного плеча 22 хромосомы локус 11.2
САТСН 22 — Cardiac defects, Abnormal facies, Thymic hypoplasia, Cleft palate, Hypocalcemia, 22q deletion – порок сердца, лицевые аномалии, гипоплазия тимуса, расщелина неба, гипокальцемия, делеция 22
FISH –fluorescent in situ hybridization — флуоресцентная гибридизация in situ
ТВХ 1 ген –Т бокс 1 ген
TREC — T-cell Receptor Excision Circles
Термины и определения
Внутривенные иммуноглобулины – препараты, содержащие преимущественно нормальный человеческий IgG. Изготовляются из пулированной плазмы тысяч здоровых доноров, с применением специальных методов очистки и вирусинактивации.
Делеция – потеря участка хромосомы
Хромосомные аберрации – изменение числа иили структуры хромосом
Микрогнатия — недоразвитие (гипоплазия) челюстных костей.
Ретрогнатия — смещение челюстной кости в дорзальном направлении (кзади)
Гипертелоризм — увеличенное расстояние между глазами
TREC – кольцевые фрагменты ДНК, образующиеся при развитии Т лимфоцитов в тимусе, в частности, в процессе формирования Т клеточного рецептора. Их концентрация в крови отражает эффективность тимопоэза. Используется для скрининга Т клеточных иммунодефицитов.
Трансплантация гематопоэтических стволовых клеток – метод лечения некоторых наследственных и приобретенных гематологических, онкологических и иммунных заболеваний, основанный на замене собственного, патологического кроветворения больного на нормальное кроветворение донора.
1.1 Определение
Синдром делеции 22-й хромосомы (синдром del 22q11) или синдром ДиДжоржи (СДД) — это совокупность морфологических, иммунологических и неврологических изменений, которые являются следствием делеции длинного плеча одной копии 22-й хромосомы — del 22q11.2 [1,2].
В классическом понятии этот синдром представляет собой комплекс симптомов, состоящий из патологии лицевого скелета (расщелины твердого неба), врожденного порока сердца, иммунодефицита вследствие гипоплазии (аплазии) тимуса и гипокальциемии, как результат гипоплазии паращитовидной железы [1,3].
Как ни один другой синдром, синдром del 22q11.2 вариабелен в количестве признаков и степени их выраженности, что и объясняет тот факт, что этот синдром в литературе имеет порядка десятка различных названий, включая синдром ДиДжорджи, САТСН 22, велокардиофациальный сидром, Шпринтцена синдром, Кайлера синдром, синдром лицевых и конотрункальных аномалий и т.д.[1,3,4].
1.2 Этиология и патогенез
В основе заболевания лежит нарушение формирования органов, происходящих их третьей жаберной дуги (нижняя часть лицевого скелета, тимус, паращитовидная железа, верхние отделы сердца и магистральных сосудов). Цитогенетические и молекулярные исследования показали, что делеция 22q11.
2 является ведущей причиной СДД и возникает спорадически более чем в 90% случаев [5,6,7]. В 10% случаев делеция наследуется от одного из родителей, так как наследование происходит аутосомно- доминантным путем [1,4].
В редких случаях синдром является проявлением перестроек других хромосом, а также мутации гена ТВХ1 [4].
Анализ ДНК пациентов с СДД хромосомы выявил, что в 85-90% случаев выпадающий участок является одним и тем же. Дефект находится между D22S427 на 22q11.21 и D22S801 на 22q11.23. В этом участке локализовано не менее 40 генов, что составляет около 3 млн пар нуклеиновых оснований.
В 10-12% случаев встречаются более короткие делеции, которые составляют 1,5-2 млн парных оснований. Было описано несколько пациентов с синдромом делеции 22-й хромосомы, имеющих делеции за пределами наиболее часто выпадающих участков [5,6].
Результаты проведенных исследований свидетельствуют о том, что степень выраженности фенотипа не коррелирует с размером делеции, т.е. пациент с потерей 1,5 млн парных оснований может иметь такой же по тяжести фенотип, как и с делецией в 3 млн парных оснований [2,5].
Кроме того, было замечено, что вариабельность фенотипических проявлений варьирует как внутри одной семьи, так и между семьями, несмотря на идентичные участки делеции [5].Делеция вызывает выпадение участка, включающего ген ТВХ, ген фактора транскрипции, участвующего в развитии фарингеальных дуг [5,6]. Эти изменения, в свою очередь, ведут к нарушению формирования сердца и магистральных сосудов, иммунологическим изменениям, расщеплению нёба и верхней губы, гипопаратиреоидизму, задержке умственного развития.
Несмотря на то, что ТВХ1, без сомнения, является главным геном, формирующим фенотип при синдроме делеции 22-й хромосомы, в результате исследований были выявлены и другие гены, недостаточная экспрессия которых может играть роль в формировании фенотипических проявлений [6,7].
Учитывая результаты работ по выяснению молекулярных основ заболевания, ясно, что в формировании фенотипа играет роль комплексное нарушение экспрессии и взаимодействия генов, их модификаторов и других составляющих, что приводит к дальнейшему нарушению эмбрио- и органогенеза [4,5,7].
Соответственно, при отсутствии или нарушении функции и экспрессии генов и дальнейших процессов происходит формирование пороков развития, характерных для СДД [1,5].
1.3 Эпидемиология
Синдром делеции 22-й хромосомы — одна из самых частых делеций среди других хромосомных аберраций в человеческом геноме, по частоте она уступает лишь синдрому Дауна, трисомии по 21-й хромосоме. Частота встречаемости варьирует от 1:4000 до 1:6000 новорожденных [1,2,3].
Не наблюдается ни половой, ни этнической предрасположенности к данному синдрому. Большинство пациентов с СДД имеют патологию лицевого скелета и врожденный порок сердца и развивают гипокальциемию вскоре после рождения [6].
Пациенты, не имеющие данных симптомов, зачастую диагностируются в раннем возрасте, и правильный значительно запаздывает.
1.4 Кодирование по МКБ-10
Другие иммунодефициты (D84):
D84.1 – Синдром ДиГеорга.
1.5 Классификация
Исторически сложилось, что в литературе часто используется разделение синдрома на полный и неполный (частичный) [1,3,5]:
- Термин «Полный синдром ДиДжорджи» использовался у пациентов, имеющих полный спектр типичных проявлений, включая выраженный иммунодефицит.
- Термин «Частичный синдром ДиДжорджи» использовался у пациентов, если они имели лишь некоторые типичные признаки, особенно без проявлений выраженного иммунодефицита. Частичный синдром делеции 22-й хромосомы в значительной степени превалирует по количеству в сравнении с полным.
2.1 Жалобы и анамнез
Спектр клинических проявлений при синдроме делеции 22-й хромосомы достаточно широк [3,6,9,11,12,14,15], поэтому жалобы и анамнез заболевания могут быть крайне разнообразными и различными по степени выраженности:
- Врожденный порок сердца представлен не менее, чем в 80% случаев. Некоторые из пороков являются более патогномоничными: прерывание дуги аорты, общий артериальный ствол и тетрада Фалло являются наиболее частыми среди данной группы детей [6,11].
- Гипокальциемия/гипопаратиреоз может проявляться судорожным синдромом при выраженном дефиците кальция в младенческом возрасте [6,12].
- Поражение носоглоточного аппарата выявлено примерно в 70% случаев и проявляется в виде велофарингеальной недостаточности, расщеплении нёба, губы, раздвоении уздечки нёба, гнусавым оттенком голоса, также описано снижение обоняния, кондуктивная и/или сен- соневральная тугоухость [6,10,13].
- Характерные черты лица (удлиненное лицо, микрогнатия или ретрогнатия, широкая переносица, мелкие зубы, асимметрия лица при плаче, опущенные вниз уголки рта, глазной гипертелоризм, низко посаженные и деформированные ушные раковины, бульбообразный кончик носа) [2,5,10].
- Иммунологические нарушения встречаются в 77% случаев. Однако инфекционные проявления вследствие дефекта иммунной системы дебютируют не с рождения. Чаще других звеньев поражается Т-клеточное звено, что проявляется предрасположенностью к грибковым заболеваниям, пневмоцистной инфекции, некоторым бактериальным и вирусным инфекциям [1,8,10].
- Нарушение выработки Т-клеток может предрасполагать к аутоиммунным заболеваниям. Описаны такие осложнения синдрома делеции 22-й хромосомы, как ЮРА, ХТП, АИГА, ВЗК, болезнь Грейвса, аутоиммунный увеит, бронхиальная астма [1,9,10,14].
- Задержка физического развития нередко наблюдается у пациентов с синдромом делеции 22q11.2-й хромосомы, которые несколько отличаются от стандартных таблиц [1,2,6,10].
- Задержка речевого и психомоторного развития наблюдается у 70—90% и проявляется с возрастом, однако тестирование пациентов с задержкой развития имеет смысл только в случаях сочетании с другими признаками [2,10,15].
2.2 Физикальное обследование
Физическое развитие большинства пациентов низкое и дисгармоничное по весу [1,10].
Стигмы дисэмбриогенеза широко вариабельны и не являются патогномоничными, однако чаще других признаков обращают на себя внимание глазной гипертелоризм, бульбообразный кончик носа и низко посаженные и или деформированные ушные раковины [6,10,13].
Могут проявляться признаки дыхательной и сердечной недостаточности [2,11]. Могут встречаться пороки развития дыхательной, пищеварительной, костно-мышечной и других систем.
Задержка умственного и речевого развития встречается у подавляющего числа пациентов с данным синдромом [2,3,15].
2.3 Лабораторная диагностика
- Рекомендуется клинический анализ крови [2,3].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 3)
- Рекомендуется исследование уровня ионизированного кальция, паратиреоиного гормона [12].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2)
- Рекомендуется исследование уровня гормонов щитовидной железы [12].
Уровень убедительности рекомендаций С (уровень достоверности доказательств – 3)
- Рекомендуется исследование уровня сывороточных иммуноглобулинов и клеточного иммунитета, включая определение количества наивных Т-лимфоцитов [1,10,16,17].
Уровень убедительности рекомендаций В (уровень достоверности доказательств – 2)
Хромосомные аномалии плода — причины, диагностика, прогнозы

Хромосомные патологии плода – это тяжелые сбои развития эмбриона, все чаще встречающееся у беременных. Большинство из них можно диагностировать в первом триместре. Лечению они не поддаются. Родители имеют право выбора и могут заранее узнать о предстоящих трудностях. Причины и проявление таких нарушений подробно описано в данной статье.
Что это такое
Хромосомные аномалии – это генетические заболевания, которые возникают при изменении количества или структуры в цепочке ДНК. Часто наблюдается трисомия 21 хромосомы.
Заболевание встречается у 1 из 700 новорожденных. Характерным является отклонение умственного развития, специфические признаки со стороны внешности. Особенно выделяется лицо, глаза.
Вторая распространенная аномалия 13, 18 хромосом. Такие отклонения намного опаснее, чем синдром Дауна. Часто дети с такими нарушениями не выживают.
Новорожденные имеют выраженные дефекты развития со стороны внешности и внутренних органов. Данные патологии проявляются в серьезных умственных отклонениях.
В медицине описано около 300 разных хромосомных нарушений.
Причины
В первую очередь при планировании ребенка, врач должен провести опрос будущих родителей и узнать о наличии наследственных заболеваний. Также большое значение имеют условия и уровень жизни. Эти факторы являются ведущими в появлении генетических мутаций.
В группу риска входят будущие мамы и папы:
- возраст которых старше 35 лет;
- с наличием таких заболеваний у кровных родственников;
- проживающие в экологически загрязненных регионах;
- с вредными условиями работы.
Такие факторы повышают вероятность развития хромосомных патологий особенно при наличии генетической предрасположенности. Если отклонения обнаруживают своевременно, врачи не советуют паре рожать.
При наступлении беременности оценивают степень поражения эмбриона, шансы на его выживаемость и дальнейшую жизнь.
Как распознать
Признаки хромосомной аномалии у плода считаются условными, поскольку до конца не изучены. К ним относятся:
Признаки являются неоднозначными и могут быть нормой, учитывая особенности организма будущей мамы и малыша. Точные результаты можно получить только с помощью анализов крови, УЗИ.
Методы диагностики
Информативным методом диагностики считается скрининг 1 триместра. Его проводят в 12 недель. Он состоит из УЗИ обследования и анализа крови.
Важно понимать, что анализ достоверный, но не может показать 100% положительный или отрицательный результат. Поэтому гарантии не дает никто.
На таком этапе врач рассчитывает вероятность. При этом учитывает возраст родителей и их анамнез.
Второй скрининг не предоставляет такой информативности. Более точные сведения можно получить инвазивными методами:
Их проводят с целью определения кариотипа. С помощью изучения биоматериала и определяют хромосомные аномалии плода.
Расшифровка и риски
Риски ХА у плода должен определить врач после полученных результатов. Обычно расшифровкой занимается гинеколог, но возможно понадобится консультация генетика. Окончательное решение выносится на основании исследований крови и ультразвуковой диагностики.
Результаты УЗИ
В процессе УЗИ оценивают строение будущего человека. При ХА обнаруживают соответствующие признаки. Ими являются:
- длина носовой кости;
- строение внутренних органов (особенно мочевыделительной системы);
- толщина воротникового пространства.
При наличии нарушений визуально заметны задержки в развитии эмбриона. Это сопровождается дисфункцией кровотока и плаценты.
Показатели крови
В этом случае оценивают результаты скринингов, анализов на определения уровня гормонов. Расшифровка осуществляется в пропорциях количества гормонов. К примеру – 1:100 означает высокую вероятность, 1:1000- среднюю, 1:100000 – низкую.
Прогнозы
Родители, которые столкнулись с хромосомными аномалиями у ребенка предстоит нелегкий выбор. Данные отклонения, как и любые генетические заболевания, являются не излечимыми. Медицина может предложить только искусственное прерывание беременности.
Такое решение принять нелегко, но важно здраво оценивать все риски. Перед этим важно уточнить такую информацию у врача:
- вид патологии, которую диагностировали;
- возможные последствия;
- вероятность угрозы выкидыша и замирания;
- сколько лет живет ребенок с определенными отклонениями.
Ответы на вопросы помогут определить, готова ли семья воспитывать ребенка-инвалида. Это поможет принять решение.
К примеру, дети с синдромом Дауна очень светлые и добрые. Если обнаружилась 21 хромосома при беременности, в любом случае есть право на выбор.
Возможные заболевания
Хромосомные аномалии плода могут стать причиной самых опасных последствий. Часто это связано с внешними уродствами, поражением ЦНС. Многое зависит от того, какой вид нарушения распространился. Например, изменения повлияли на их количество или структуру.
Нарушение числа хромосом
К ним относятся следующие синдромы.
- Дауна – патология 21 пары, сопровождается слабоумием, нарушением физического развития, характерной внешностью лица.
- Патау – отклонения в 13 хромосоме, тяжелая аномалия, для которой характерно много пороков, дети обычно выживают до года.
- Эдвардса – нарушение в 18 паре, причина обычно скрывается в возрасте будущих родителей, большинство детей умирают до 3 месячного возраста.
Изменения в количестве половых хромосом
К таким отклонениям относятся:
- синдром Шерешевского-Тернера – характерным является неправильное формирование половых желез, которое наблюдается в основном у девушек (это связано с отсутствием или дефектом Х-хромосомы);
- полисомии по Х- или Y- хромосоме – это разные нарушения, которые сопровождаются снижением интелекта;
- сидром Клайнфельтера – аномалия связана с Х-хромосом у мальчиков, которые выжили после родов, но имеют характерный внешний вид.
Полиплоидия
Тяжелая аномалия, которая в большинстве случаев заканчивается летальным исходом еще до момента рождения. Родителям не остается выбора, как прервать беременность. Поскольку детки не выживают.
Что делать
Анализ на хромосомные патологии, их риск следует исключать до планирования беременности. При вероятности генетических болезней, необходимо пройти консультацию генетика. Специалист определяет возможность родить и выносить здорового малыша.
В противном случае следует ожидать непростого выбора и решения провести искусственный аборт. Но многие не отказываются от детей-инвалидов и счастливо их воспитывают.
Материал подготовлен
специально для сайта kakrodit.ru
под редакцией врача-диагноста Васильевой О.С.
Хромосомные патологии при беременности
:
Хромосомные патологии при беременности, к сожалению, совсем не такая редкость, как хотелось бы.
Но, есть и хорошая новость – многие хромосомные патологии, по крайней мере, самые часто встречающиеся, можно диагностировать с помощью различных тестов еще на ранних сроках беременности.
Конечно же, вылечить это невозможно даже при самой ранней диагностике, но у будущих родителей хотя бы появляется выбор – готовы ли они к рождению особенного ребенка, или примут решение прервать беременность.
Если родители в любом случае решают оставить малыша, какое заболевание не было бы диагностировано, эта информация дает возможность максимально подготовиться к жизни после родов, обдумать, как и где они смогут лечить ребенка, будут теоретически знать, какие особенности ухода нужно иметь в виду и прочие важные нюансы.
Для начала посмотрим, что такое хромосомные патологии, какие они бывают, и что такое вообще хромосомы.
Хромосомы представляют собой палочковидные структуры в середине каждой клетки в организме. Каждая клетка имеет 46 хромосом, сгруппированных в 23 пары. Когда хромосома является ненормальной, это может вызвать проблемы со здоровьем в организме. Аномальные хромосомы чаще всего образуются в результате ошибки во время деления клетки.
Аномалии хромосом часто происходят из-за одного или нескольких факторов:
- Ошибки при делении половых клеток (мейоз)
- Ошибки при делении других клеток (митоз)
- Воздействие веществ, вызывающих врожденные дефекты (тератогены)
Мейоз
Мейоз — это процесс, при котором половые клетки делятся и создают новые половые клетки с половиной числа хромосом. Сперматозоиды и яйцеклетки — это половые клетки. Мейоз — это начало процесса развития ребенка. Когда сперматозоид оплодотворяет яйцеклетку, соединение приводит к зачатию ребенку с 46 хромосомами – это в случае нормы.
Но если мейоз не происходит нормально, у ребенка может быть лишняя хромосома (трисомия) или недостающая хромосома (моносомия). Эти проблемы могут привести к потере беременности, или они могут вызвать проблемы со здоровьем у ребенка.
Женщина в возрасте 35 лет и старше имеет более высокий риск рождения ребенка с хромосомной аномалией. Это потому, что ошибки в мейозе могут быть более вероятны в результате процесса старения. Женщины рождаются уже со всеми яйцеклетками в яичниках, но они начинают созревать в период полового созревания. Если женщине 35 лет, яйцеклеткам в яичниках также 35 лет.
Вас могут направить на анализ крови на хромосомные патологии при беременности, если вы беременны и старше 35 лет. В мужском организме процессы образования сперматозоидов продолжаются на протяжении всего репродуктивного периода. Поэтому, возраст не сильно увеличивает риск хромосомных нарушений у возрастных отцов.Но более новые исследования предполагают, что редкие отклонения все же происходят.
Митоз — это деление всех других клеток в организме. Митоз приводит к тому, что количество хромосом в клетке удваивается до 92, а затем клетка делится пополам и количество хромосом также делится пополам — по 46.
Этот процесс постоянно повторяется в клетках по мере роста ребенка. Митоз продолжается на протяжении всей вашей жизни.
Он заменяет клетки кожи, клетки крови и другие типы клеток, которые повреждены или естественным образом погибают.
Во время беременности может возникнуть ошибка в митозе. Если хромосомы не делятся на равные половины, новые клетки могут иметь дополнительную хромосому (всего 47) или недостающую хромосому (всего 45).
Тератоген
Тератоген — это то, что может вызвать или повысить риск врожденного дефекта у ребенка. Это то, с чем может столкнуться мать во время беременности. Тератогены включают в себя:
- Некоторые лекарства
- Наркотики
- Алкоголь
- Табак
- Токсичные химикаты
- Некоторые вирусы и бактерии
- Некоторые виды излучения
Каждая из наших хромосом имеет характерную структуру. Исторически ученые использовали технику окрашивания, которая окрашивает хромосомы в полосатый рисунок. Эти шаблоны полос облегчают идентификацию каждой из наших отдельных хромосом, и благодаря этому можно составить визуальное изображение кариотипа. Любое отклонение от нормального кариотипа известно как аномалия хромосомы.
Половина всех самопроизвольных абортов происходит из-за хромосомных нарушений.
Самые серьезные хромосомные расстройства вызваны потерей или приобретением целых хромосом. Какие это могут быть заболевания:
- Трисомия 21 хромосомы – синдром Дауна (15 на 10 000)
- Трисомия 18 хромосомы – синдром Эдвардса (3 на 10 000)
- Трисомия 13 хромосомы – синдром Патау (2 на 10 000)
- Моносомия Х-хромосомы – синдром Шерешевского-Тернера (2 на 10 000)
- Кариотип XXY – синдром Клайнфельтера (10 из 10 000)
- Кариотип XXX (возможно и большее количество Х-хромосом, например, ХХХХХ) – синдром «суперженщины» (10 на 10000)
- Кариотип XYY (возможно большее количество Y-хромосомы – XYYY) – синдром «супермужчины» (10 на 10000)
- Структурные нарушения могут принимать несколько форм:
- Делеция – утрата части хромосомы, и вследствие этого, части генов, ответственных за те или иные функции в организме
- Дупликация – мутация, вызывающая повторение участка хромосомы, что приводит к дополнительному генетическому материалу.
- Транслокация – мутация, вызывающая перемещение одной части хромосомы в другую часть хромосомы (внутрихромосомно) или в другую хромосому вообще (межхромосомно). Есть два ключевых типа: обратный: сегменты из двух разных хромосом обмениваются частями, и второй — Робертсонский: вся хромосома прикрепляется к другой.
- Инверсия: мутация, какой-то участок хромосомы оказывается повернутым на 180о .
- Кольцевая хромосома – когда в обоих плеча хромосомы отсутствуют конечные фрагменты и они замыкаются, образуя круг
- Изохромосомия – когда в хромосоме фрагменты ДНК повторяются в обоих плечах
Структурные аномалии – это когда большие участки ДНК отсутствуют или лишние, при нормальном количестве хромосом.
Сбалансированные структурные аномалии включают перестройку генетического материала, но без общего прибавления или потери генетического материала, то есть «сумма» неизменна. Например, инверсии и транслокации.
Несбалансированные структурные отклонения связаны с получением или потерей генетического материала – остальные варианты структурных нарушений. Даже крошечные несбалансированные структурные аномалии могут повлиять на многие гены и, следовательно, оказать серьезное влияние на человека.
Чаще всего в практике встречаются транслокации и делеции, нежели другие виды структурных аномалий хромосом.
Несбалансированные структурные отклонения могут быть такими:
- Синдром Вольфа — Хиршхорна – делеция короткого плеча хромосомы 4 (1 на 50 000)
- Синдром кошачьего крика – делеция короткого плеча хромосомы 5 (1 на 50 000)
- Синдром WAGR – микроделеция короткого плеча хромосомы 11 (1 на 500 000 до 1 миллиона)
- Микроделеция Прадера-Вилли / Ангельмана с короткого плеча хромосомы 15 (1 на 15 000)
- Микроделеция Ди-Джорджи из длинного плеча хромосомы 22 (1 в 4000)
Детальная характеристика хромосомной патологии плода при беременности, частой причины врожденных патологий и потери беременности, имеет решающее значение для выяснения генов для развития плода человека.
Среди всех хромосомных аномалий, связанных с самопроизвольным прерыванием беременности, наиболее часто обнаруживаются хромосомные анеуплоидии, которые включают изменение числа копий всей хромосомы, например, трисомию 21, трисомию 13, трисомию 18 и моносомию X, и их встречаемость значительно возрастает с возрастом матери. Хромосомные структурные аномалии, возникающие из-за изменения структуры или частей хромосомы, например, делеции и дупликации, встречаются реже, чем хромосомная анеуплоидия, но скорость их обнаружения значительно улучшилась благодаря применению цитогенетических методов на основе микрочипов.
Рассмотрим немного детальнее некоторые заболевания, которые вызваны нарушениями со стороны количества или качества хромосом.
Пожалуй, самым распространенным заболеванием, о котором знают все, является синдром Дауна. При этом заболевании лишняя 21 хромосома вызывает пороки развития, но, как правило, они совместимы с жизнью, а в реалиях современного уровня медицины серьезные проблемы со здоровьем коррегируются, чем улучшаются прогнозы относительно качества жизни и продолжительности жизни.
Синдром Эдвардс
Синдром Эдвардса – возникает вследствие дополнительной 18 хромосомы. Такие детки рождаются с множественными врожденными пороками развития, причем это чаще девочки. Такие дети маловесные, при том что беременности донашиваются до нормального срока.
Как правило, для синдрома характерны, прежде всего, аномалии строения черепа – он имеет вытянутую форму, и лицевой череп также патологически развивается. Нижняя челюсть недоразвита, может быть расщелина неба и верхней губы, узкие глазные щели, деформированные, низкорасположенные ушные раковины.
Иногда может отсутствовать слуховой проход. Грудная клетка более широкая и короткая, чем у здоровых детей. Характерны и изменения конечностей. Кроме того, аномалии и со стороны внутренних органов – пороки сердца, сосудов, нарушения со стороны формирования мозговых структур, нарушение мышечного тонуса.
К сожалению, несмотря на любое лечение и уход, такие дети умственно не сохранны, интеллект сильно нарушен.Продолжительность жизни этих детей небольшая – от 3 месяцев до года, но при легких формах заболевания ребенок может прожить несколько лет.
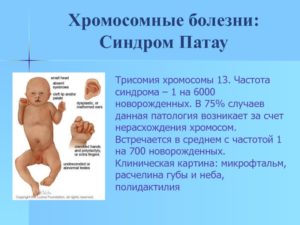
Синдром Патау
Синдром Патау – трисомия 13 хромосомы. Это очень тяжелая хромосомная аномалия, которая приводит к рождению глубоко инвалидизированных детей. Такие дети также рождаются обычно доношенными, но умеренно маловесными.
При беременности характерно многоводие, которое встречается в половине случаев при этой хромосомной патологии.
Из аномалий развития чаще всего встречаются: уменьшение объема мозга – микроцефалия, узкие глаза, возможны аномалии развития глаз в виде циклопии, уменьшении размеров глазных яблок, широкое основание носа, расщелины неба и губы, деформированные ушные раковины, могут быть лишние пальцы на кистях рук и стопах, пороки развития внутренних органов – от порока сердца до удвоения селезенки, почки имеют аномальное строение, половые органы также дефектны. Умственно такие дети не сохранны. Почти все дети с этим синдромом умирают в первый год жизни, но иногда могут жить несколько лет.
Синдром кошачьего крика
Синдром кошачьего крика – патология, которая вызывается делецией короткого плеча 5 хромосомы. Кариотип ребенка при этом — 46 XX или XY, 5р-. При этом выраженность проявлений синдрома зависит не от величины дефекта в хромосоме, а от отсутствия определенного маленького участка хромосомы.
Бывают также случаи мозаицизма, тогда проявления менее выражены. Особенностью детей с таким синдромом, что позволяет предположить патологию еще в родильном доме – характерный «мяукающий» плач ребенка, что происходит из-за аномального строения гортани.
Эта особенность обычно исчезает после первых 12 месяцев жизни. Также характерны маловесность, отставание в развитии, снижение мышечного тонуса, лунообразное лицо и широко расставленные глаза. С синдромом кошачьего крика девочки рождаются немного чаще, чем мальчики.
Кроме того, у таких детей часто бывает микроцефалия, пороки сердца, гипертелоризм, пороки внутренних органов и опорно-двигательной системы. При квалифицированном уходе, медицинском сопровождении прогнозы для жизни достаточно хорошие, но задержка психомоторного и физического развития имеет место.
В любом случае, само по себе заболевание коррекции не поддается, но улучшить качество жизни таких детей вполне возможно.
Cиндром WAGR
Еще один довольно редкий синдром – синдром WAGR. Название этого синдрома – аббревиатура от наиболее частых проявлений заболевания: W – опухоль Вильямса, A – аниридия, G – аномалии со стороны репродуктивных органов, R – отставание в умственном развитии.
Аниридия – это отсутствие радужной оболочки, и обычно сопровождается и другими аномалиями строения и развития органа зрения. Опухоль Вильямса – это нефробластома – очень злокачественная опухоль, которая характерна для детей до 5 лет, пол не важен.
Не всегда при этом генетической патологии наблюдаются сразу все эти признаки, и иногда проявления ограничены не только ними. Иногда этот синдром вообще обнаруживается при обследовании по поводу, например, аниридии.Изолированное проявление какой-то из составляющих синдрома может и не вызвать подозрения на генетическую патологию, или стать явным уже в более старшем возрасте. Этот синдром не передается по наследству и является следствием спонтанной мутации, поэтому такие дети могут рождаться и у абсолютно здоровых родителей, как впрочем, при почти всех хромосомных аномалиях.
Многие хромосомные патологии могут иметь схожие проявления, и не всегда фенотипически можно сразу точно поставить диагноз. Для этого необходимо провести анализ кариотипа, чтобы иметь возможность давать прогнозы относительно здоровья и жизни таких детей