Трисомия 10 хромосомы
Анеуплоидия

Анеуплоиди́я (др.-греч. ἀν- — отрицательная приставка + εὖ — полностью + πλόος — кратный + εἶδος — вид) — изменение кариотипа, при котором число хромосом в клетках не кратно гаплоидному набору (n).
Отсутствие в хромосомном наборе диплоидного организма одной хромосомы называется моносомией (2n-1); отсутствие двух гомологичных хромосом — нуллисомией (2n-2); наличие дополнительной хромосомы называется трисомией (2n+1) . Анеуплоидия возникает в результате нарушения сегрегации хромосом в митозе или мейозе.
Анеуплоидия вызывает у человека некоторые наследственные синдромы. Анеуплоидия по аутосомам нарушает нормальное эмбриональное развитие и является одной из основных причин спонтанных абортов[1]:1. Анеуплоидия характерна для опухолевых клеток, особенно для клеток сóлидных опухолей[2].
Патологический фенотип при анеуплоидии формируется из-за нарушения дозового баланса генов, при моносомии дополнительный негативный вклад оказывает гемизиготное состояние генов моносомной хромосомы[3] :1.
Врождённая анеуплоидия может возникнуть, если в анафазе I мейоза гомологичные хромосомы одной или нескольких пар не разойдутся.
В этом случае оба члена пары направляются к одному и тому же полюсу клетки, и тогда мейоз приводит к образованию гамет, содержащих на одну или несколько хромосом больше или меньше, чем в норме. Это явление известно под названием нерасхождение.
Когда гамета с недостающей или лишней хромосомой сливается с нормальной гаплоидной гаметой, образуется зигота с нечётным числом хромосом: вместо каких-либо двух гомологов в такой зиготе их может быть три или только один.Зигота, в которой количество аутосом меньше нормального диплоидного, обычно не развивается, но зиготы с лишними хромосомами иногда способны к развитию. Однако из таких зигот в большинстве случаев развиваются особи с резко выраженными аномалиями.
Формы анеуплоидии[ | ]
Схематическое изображение кариотипа мужчины, страдающего синдромом Дауна. Нерасхождение хромосом 21 в мейозе привело к трисомии по этой хромосоме.
Детекция трисомии по хромосоме 7 и 19 в культивируемых in vitro нейральных клетках-предшественниках при помощи дифференциального G-окрашивания и FISH-метода
По типу вовлечённых хромосом выделяют анеуплоидию половых хромосом и аутосомную анеуплоидию.
Анеуплоидия по половым хромосомам характеризуется значительно более мягкими фенотипическими проявлениями, чем анеуплоидия по аутосомам, так как в отношении Х-хромосомы работает механизм дозовой компенсации, а Y-хромосома несёт малое количество генов и добавочная Y-хромосома незначительно нарушает дозовый баланс.
По числу вовлечённых хромосом анеуплоидию классифицируют как нуллисомию при отсутствии пары гомологичных хромосом, моносомию при отсутствии одной из пары гомологичных хромосом, трисомию при наличии добавочной хромосомы. Для половых хромосом у человека описаны случаи тетрасомии (48XXXX, 48XXYY, 48XXXY, 48XYYY) и пентасомии (49XXXXX, 49XXXXY, 49XXXYY, 49XYYYY, 49XXYYY)[4].
Моносомия[ | ]
Последствия моносомии являются, как правило, более тяжёлыми по сравнению с трисомией. В случае моносомии негативный эффект анеуплоидии обусловлен не только нарушенным дозовым балансом, но и гемизиготным состоянием генов, находящихся на хромосоме, не имеющей пары. Моносомии по аутосомам у человека являются эмбрионально летальными.
Моносомия по Х-хромосоме у женщин приводит к синдрому Шерешевского-Тёрнера.
Моносомия по Y-хромосоме является летальной перестройкой и несовместима с живорождением.
В случае обширной делеции в какой-либо хромосоме иногда говорят о частичной моносомии. Примером может служить синдром кошачьего крика, причиной которого является утрата части короткого плеча хромосомы 5.
Трисомия[ | ]
Трисомия — это наличие трёх гомологичных хромосом вместо пары (в норме). Причиной подавляющего большинства трисомий у человека являются ошибки расхождения хромосом при оогенезе, причём наибольший вклад дают ошибки в мейозе I по сравнению со вторым мейотическим делением. Вероятность трисомий у потомства повышается с возрастом матери[1]:2.
Наиболее часто встречающейся у человека является трисомия по 16-й хромосоме (более одного процента случаев беременности), следствием этой трисомии является спонтанный выкидыш в первом триместре беременности.
Единственной жизнеспособной трисомией по аутосоме у человека является трисомия по хромосоме 21, вызывающая синдром Дауна.
Трисомики по хромосомам 13 (синдром Патау) и 18 (синдром Эдвардса) могут дожить до рождения, но характеризуются значительными нарушениями развития и ранней постнатальной смертностью.
Трисомии по другим аутосомам приводят к ранней эмбриональной летальности. Характерно, что хромосомы 13, 18 и 21 являются хромосомами, занимающие три последних места по числу генов среди аутосом[3]:2.
Частота новорождённых с трисомией по 21 хромосоме в европейских странах в 1990—2009 годах составляло 11.2 случаев на 10 000 новорождённых, по 18 хромосоме — 1.04 случаев на 10 000, по 13 хромосоме — 0.48 случая на 10 000[5].
Анеуплоидии половых хромосом[ | ]
- 45X, синдром Шерешевского — Тёрнера
- 47XXX (женщины без фенотипических особенностей, у 75 % наблюдается умственная отсталость различной степени, алалия. Нередко недостаточное развитие фолликулов в яичниках, преждевременное бесплодие и ранний климакс (необходимо наблюдение эндокринолога). Носительницы ХХХ плодовиты, хотя риск спонтанных абортов и хромосомных нарушений у потомства у них несколько повышен по сравнению со средними показателями; частота проявления 1:700)
- 47XXY, синдром Клайнфельтера (мужчины, обладающие некоторыми вторичными женскими половыми признаками; бесплодны; яички развиты слабо, волос на лице мало, иногда развиваются молочные железы; обычно низкий уровень умственного развития)
- 47XYY (мужчины высокого роста с различным уровнем умственного развития)
Тетрасомия и пентасомия[ | ]
Примерами тетрасомии и пентасомии у человека могут служить кариотипы 48XXXX, 48XXYY, 48XXXY, 48XYYY, 49XXXXX, 49XXXXY, 49XXXYY, 49XYYYY и 49XXYYY. Такие случаи встречаются чрезвычайно редко с частотой 1:18 000—1:100 000[6]. Как правило, с нарастанием количества «лишних» хромосом увеличивается тяжесть и выраженность клинических симптомов.
См. также[ | ]
- Плоидность
- Хромосомные перестройки
- Хромосомные болезни
Примечания[ | ]
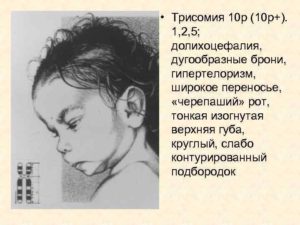
Дистальная трисомия 10q. Симптомы, диагностика, лечение
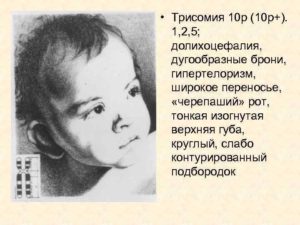
Дистальная трисомия 10q является крайне редким хромосомным расстройством, при котором дистальный конец длинного плеча (q) в хромосоме 10 (10q) имеется в каждой клетке в трех экземплярах (трисомия), а не в двух.
Это расстройство характеризуется необычно медленным ростом до и после рождения, аномально слабым мышечным тонусом (гипотония), умственной отсталостью и задержками в психомоторном развитии.
Дети, с дистальной трисомией 10q, также могут иметь отличительные пороки развития головы и лица (черепно-лицевые пороки), дефекты рук и / или ног, скелетные аномалии, пороки сердца, почек и / или легких.
Диапазон и тяжесть проявлений могут варьироваться от случая к случаю, в зависимости от точной длины и расположения дублируемой части хромосомы 10. В большинстве случаев, дистальная трисомия 10q связана с сбалансированной хромосомной транслокацией у одного из родителей.
Дистальная трисомия 10q. Эпидемиология
Дистальная трисомия 10q является чрезвычайно редким, но хорошо определяемым хромосомным расстройством, которое, способно развиваться как у лиц мужского так и женского пола, примерно с одинаковой частотой. Более 35 случаев было зарегистрировано в медицинской литературе с 1974 году. Многие из симптомов и физических особенностей, связанных с этим расстройством, очевидны при рождении.
Дистальная трисомия 10q. Причины
Дистальная трисомия 10q является крайне редким хромосомным расстройством, при котором дистальная часть длинного плеча в хромосоме 10 имеется в трех экземплярах. Дублирование дистальной части хромосомы 10q отвечает за развитие симптомов и физических особенностей, которые характеризуют дистальную трисомию 10q.
Диапазон и тяжесть симптомов зависят от точной длины и расположения продублированной части. Длинна этой аномальной части может сильно отличаться у разных лиц. Согласно медицинской литературе, исследователи подозревают, что дублирование полос 10q25 и 10q26 имеет решающее значение в выражении характерных особенностей, связанных с этим расстройством.
Очень часто, аномалии сердца и почек связаны с дублированием полосы 10q24. В более чем 90% зарегистрированных случаев, трисомия 10q была связана с сбалансированной хромосомной транслокацией у одного из родителей.В некоторых случаях, дистальная трисомия 10q может быть связана с спонтанными (De Novo) генетическими изменениями (мутациями), которые происходят по неизвестным причинам.
Дистальная трисомия 10q. Симптомы и проявления
Ребенок с трисомией 10q
Физические проявления, связанные с дистальной трисомией 10q, могут сильно варьироваться в диапазоне и тяжести, а также в зависимости от точного размера и расположения продублированной части хромосомы 10.
Тем не менее, в большинстве случаев, расстройство характеризуется слабой или тяжелой умственной отсталостью, отличительными пороками развития головы и лица (черепно-лицевые пороки), дефектами рук и / или ног, скелетными аномалиями, пороками сердца, почек и / или легких.
Важно отметить, что практически все младенцы, с этой хромосомной аномалией, не будут иметь сразу все аномалии, которые мы перечислим ниже.
В большинстве случаев, дистальная трисомия 10q характеризуется аномально медленным ростом до и после рождения. Кроме того, большинство детей имеют мягко или серьезно уменьшенный мышечный тонус (гипотония). Некоторые дети также могут иметь некоторые аномалии конечностей. Младенцы и дети с дистальной трисомией 10q имеют умственное отставание и глубокие задержки в психомоторном развитии.
Кроме того, младенцы и дети, с этим расстройством, имеют характерные пороки развития головы и лица (черепно-лицевая область).
Такие нарушения могут включать в себя аномально маленькую голову (микроцефалия) с высоким, широким лбом, круглым, слегка уплощенным лицом, выделяющимися скулами и / или с низкорасположенными, деформированными ушами.
Дети также могут иметь маленький нос с загнутыми ноздрями (антеверсия ноздрей), необычно маленькую нижнюю челюсть и / или, в некоторых случаях, волчью пасть.
Дополнительные проявления могут включать в себя необыкновенно тонкие, высокие и арочные брови, опущение верхнего века (птоз), блефарофимоз и / или вертикальные складки кожи, которые могут охватывать внутренние углы глаз. В некоторых случаях, у детей могут быть широко расставленные глаза. Кроме того, у некоторых младенцев и детей, глаза могут быть аномально небольшими (микрофтальмия) из-за уменьшенного диаметра роговицы.
В некоторых случаях, дети и младенцы с дистальной трисомией 10q, также имеют характерные пороки развития рук и / или ног. Такие нарушения могут включать в себя постоянно согнутые пальцы в кулак и / или перекрытие некоторых пальцев, необычно большое расстояние между большим и вторым пальцами ног и / или ненормальное позиционирование ног.
Многие младенцы и дети, с этим расстройством, могут иметь дополнительные скелетные аномалии, например аномально тонкие ребра, 11, а не 12 пар ребер, аномально короткую шею, кифосколиоз и / или ненормальные депрессии грудины. Дети могут проявлять аномальные задержки в развитии костей.
Примерно половина детей, с дистальной трисомией 10q, имеют дефекты сердца, которые присутствуют при рождении, нарушения в дыхательной функции и / или пороки развития почек.Симптомы и проявления, связанные с врожденными пороками сердца, могут сильно варьироваться в зависимости от конкретного характера, размера и расположения анатомического дефекта.
В некоторых случаях, сопутствующие симптомы и физические проявления могут включать в себя одышку из-за неспособности сердца эффективно перекачивать кровь (сердечная недостаточность), быстрая утомляемость, цианоз кожи и слизистых оболочек и / или повышенная восприимчивость к инфекциям.
Почечные пороки, связанные с этой трисомией, могут включать недоразвитие почек (гипоплазия), развитие кист и / или ненормальных опухолей из-за накопления мочи в почках (гидронефроз). В тяжелых случаях, сердечные, дыхательные и / или почечные аномалии часто приводят к развитию опасных для жизни осложнений в течении первых нескольких лет жизни.
Дистальная трисомия 10q. Диагностика
В некоторых случаях, дистальная трисомия 10q может быть определена еще до рождения (пренатально) специализированными тестами, такими как УЗИ, амниоцентез и / или биопсия хориона.
Ультразвуковые исследования могут помочь в выявлении характерных проявлений, которые будут свидетельствовать о хромосомном расстройстве.
Дистальная трисомия 10q также может быть диагностирована и / или подтверждена после рождения на основе результатов тщательно проведенных клинических обследований, выявлении характерных физических проявлений и хромосомных исследований.
Дистальная трисомия 10q. Лечение
Лечение дистальной трисомии 10q направлено на конкретные симптомы и аномалии, которые проявляются в каждом пациенте по-своему. Лечение дистальной трисомии 10q только симптоматическое и поддерживающее.
В некоторых случаях, пациент может быть отправлен на операцию по исправлению некоторых черепно-лицевых, скелетных, сердечных, почечных и / или других пороков, которые могут быть связаны с расстройством. В таких случаях, необходимые хирургические процедуры будут зависеть от локализации и тяжести анатомических нарушений и связанных с ними симптомов.
Врачи также могут рекомендовать превентивные меры для пострадавших детей и детей, которые могут быть подвержены повышенному риску развития повторных респираторных инфекций.
Характерные признаки и методы ранней диагностики синдрома Патау

Синдром Патау представляет собой тяжёлую врождённую патологию, имеющую генетическую природу. Хромосомное заболевание встречается с частотой один случай на семь – четырнадцать тысяч новорожденных, мальчики и девочки одинаково подвержены недугу. Проявляется синдром в многочисленных аномалиях развития органов, зачастую несовместимых с жизнью.
Причины возникновения синдрома Патау
В геноме человека содержится 23 пары хромосом. Любые отклонения в их количестве или структуре ДНК приводят к аномалиям развития организма.
Состояние, когда в паре появляется лишняя третья хромосома, носит название трисомия. Генная мутация ведёт к рождению ребёнка с отклонениями в развитии.
Трисомия по 13 хромосоме (синдром, описываемый далее) означает, что именно в тринадцатой паре нуклеопротеидных структур, содержащих наследственную информацию, произошла мутация.
Синдром Патау, при котором изменён кариотип, характеризуется случайным возникновением дефекта при формировании половых клеток родителей – яйцеклетки или сперматозоида. Цитогенетические варианты развития болезни различны, но чаще всего встречаются два из них:
- Простая трисомия проявляется наличием трёх гомологичных хромосом вместо двух.
- Центрическое слияние. Представляет собой соединение двух акроцентрических хромосом, которыми и являются структуры тринадцатой пары.
Заболевание, его клинические проявления и отличие от других недугов были описаны датским медиком Расмусом Бартолином ещё в XVII веке. Обусловленность патологии дефектами генотипа была определена в шестидесятые годы XX века доктором Клаусом Патау.
Синдром Патау, причиной которого является мутация генов, имеет следующие возможные факторы развития:
- Фактор случайности. Внезапный дефект формирования хромосомного набора во время созревания гамет или образования зиготы.
- Наследственность. Если одному из родителей присущ так называемый кариотип Робертсона (описанный выше эффект центрического слияния), то с высокой долей вероятности у него родится ребёнок с трисомией 13. Точный патогенез в данном случае неизвестен. Люди с робертсоновской транслокацией ничем не отличаются от других внешне, и патология не проявляется внутренними особенностями. До рождения у них ребёнка с одним из видов хромосомных мутаций и проведения соответствующих исследований ДНК такие люди не подозревают о наличии у себя подобных аномалий.
- Радиация. Предположение о влиянии данного фактора основано на описании синдрома для тихоокеанских племён после того, как испытания ядерного оружия в регионе вызвали радиационное заражение.
- Возраст матери. Доказано, что чем старше женщина, тем больше вероятность генетических мутаций и рождения нежизнеспособных детей.
К вероятным факторам относят высокий уровень загрязнённости окружающей среды (плохая экологическая ситуация в регионе проживания родителей), половые контакты между родственниками.
Характерные признаки недуга
Синдром Патау характеризуется признаками внешнего характера:
- Небольшой (ниже нормы) вес плода – менее двух килограммов.
- Микроцефалия (череп маленьких размеров, вследствие чего голова по отношению к телу небольшая).
- Лоб низкий, скошенный.
- Узкие глазные щели, узко посаженные глаза, отсутствие части глазной оболочки.
- Запавшая переносица при широком основании носа.
- Дефекты строения ушных раковин.
- Волчья пасть – расщепление нёба и верхней губы, в данном случае чаще всего симметричное.
- Короткая шея.
- Аномалии развития конечностей – большее количество пальцев на кистях рук или на ногах.
Совокупность внутренних и внешних проявлений даёт основание для постановки диагноза. Синдрому Патау присущи симптомы, связанные с аномалиями внутренних органов:
- Центральная нервная система характеризуется такими отклонениями, как уменьшение размеров головного мозга, недоразвитие некоторых его структур и отделов. Это приводит к задержке умственного и физического развития, расстройствам психики.
- Сердечные патологии: дефекты перегородок, другие формы порока.
- Несоответствие норме диаметра сосудов по всему телу.
- Гормональные дисфункции, связанные с недоразвитием мозговых структур, ответственных за выработку гормонов.
- Нефизиологичное расположение внутренних органов.
- Со стороны репродуктивной системы – гипоплазия половых органов.
- Снижение или полное отсутствие слуха, зрения, связанное с аномалиями строения соответствующих органов.
- Мочевыделительная система характеризуется отклонениями в строении мочевого пузыря, увеличением почек.
- Врождённые пупочные грыжи.
- Фиброкистозные изменения поджелудочной железы.
Во время беременности у женщины, вынашивающей ребёнка с синдромом Патау, может быть диагностировано многоводие, роды происходят раньше времени – в 38 недель. На ранних сроках возможны выкидыши – организм матери отторгает нежизнеспособный эмбрион.
Диагностическое подтверждение патологии
Диагностика патологии может быть проведена ещё при беременности, на разных её сроках. После рождения ребёнка диагноз подтверждается на основании симптомов, наблюдающихся у новорожденного.
При беременности
Пренатальная диагностика на ранних сроках беременности позволяет определить патологию и принять решение относительно дальнейших действий.
Первый скрининг включает в себя лабораторные исследования крови, взятой из вены, и УЗИ.
Проводится такое обследование между одиннадцатой и тринадцатой неделями – именно этот срок является минимальным для того, чтобы обнаружить значимые изменения развития эмбриона.
Консультация беременных предполагает информирование женщин о вероятном прогнозе для ребёнка в случае наличия хромосомных аномалий.
Анализ крови предполагает исследование таких параметров, как ХГЧ и плазменный белок А (PAPP-A). Последний в случае развития трисомии значительно снижен.
Ультразвуковое исследование плода при наличии синдрома показывает:
- тахикардию;
- отставание в развитии;
- дефекты формирования головного мозга;
- аномальный размер мочевого пузыря.
При подозрении на трисомию проводятся дополнительные исследования: анализ клеток ворсин хориона (берётся плацентарная ткань), изучение околоплодных вод и пуповинной крови. Каждое из исследований проводится на определённом сроке, все они инвазивны, предполагают хирургическое вмешательство, в процессе которого есть угроза для матери и плода.
Современная медицина даёт женщине возможность сделать неинвазивный пренатальный тест ДНК.
Если расшифровка теста свидетельствует о низком риске развития синдрома, то использовать хирургические методы исследования нет необходимости.В случае когда тест показывает высокую вероятность патологии, необходимо всё же прибегнуть к инвазивным способам. Только их результаты могут стать основанием для искусственного прерывания беременности.
У новорожденных
Результаты обследований беременной женщины дают основание предполагать с высокой долей вероятности развитие у ребёнка описываемого синдрома. Осмотр новорожденного в данном случае подтверждает диагноз. На фото разных детей, страдающих синдромом, прослеживаются схожие признаки. При осмотре критериями являются:
- форма черепа;
- аномалии развития лица;
- патологии строения пальцев рук и ног.
Если по каким-то причинам во время вынашивания плода не были проведены соответствующие исследования, новорожденному могут быть дополнительно проведены:
- ультразвуковое исследование органов брюшной полости;
- эхокардиография;
- нейросонография.
Рекомендации по лечению
Лечение синдрома Патау, к сожалению, невозможно. Терапия сводится лишь к тому, чтобы хирургическим путём устранить некоторые дефекты внутреннего и внешнего развития, а также оказать паллиативную помощь.
Опасность недуга, прогноз и профилактика
Говорить о мерах предотвращения заболевания нецелесообразно, поскольку мутация носит внезапный характер.
Тем не менее если у родителей были выявлены отклонения (например, робертсоновская транслокация) и особенно если были случаи рождения детей с хромосомными аномалиями, перед зачатием рекомендуется пройти обследования.
В период вынашивания плода таким парам также предлагают дополнительные анализы и исследования.
Поскольку считается, что может прослеживаться взаимосвязь между развитием синдрома и возрастом матери, рекомендуется ответственно подходить к вопросу планирования семьи.
К возможным мерам первичной профилактики можно отнести проживание в районах с хорошими показателями экологии и отсутствие радиационного облучения.
Опасность синдрома в том, что множественные патологии внутренних органов бывают несовместимы с жизнью.
Прогноз для рождённых с трисомией 13 неблагоприятный. Медиана выживаемости (время, к которому умирает 50% больных) составляет 2 дня. Небольшой процент пациентов доживает до года.
При минимальных врождённых дефектах, своевременной хирургической коррекции патологий и должном уходе такие дети могут прожить до возраста 5–10 лет. Тяжёлая умственная отсталость и задержка в психическом развитии сопровождают пациентов на протяжении их жизни.
(2 3,50 из 5)
Загрузка…
Трисомия по х хромосоме
Синдром трисомии X представляет собой генетическое заболевание, наблюдаемое у женщин, характеризующееся наличием дополнительной Х-хромосомы.
Синдром Triple X также называют:
- 47, XXX
- 47, XXX Кариотип
- синдром XXX, 47
- Синдром XXX
- Трисомия X
Факты
- Встречается только у женщин
- Это не наследственное расстройство
- Синдром наблюдается у 1 из 1000 новорожденных девочек
- Некоторые случаи недиагностированы из-за отсутствия симптомов
- Приблизительно 10 процентов случаев диагностированы
Генетика синдрома Triple X
Обычно у каждого человека 46 хромосом, из которых две являются половыми, а именно X и Y. Женщины имеют две Х-хромосомы, мужчины одну Х и одну Y.
Люди, родившиеся с трисомией по х хромосоме, имеют 3 Х-хромосомы, поэтому общее количество составляет 47 из-за дополнительной Х.
Некоторые женщины с синдромом трисомии Х имеют дополнительную Х-хромосому только в некоторых клетках, которая называется 46, XX / 47, XXX мозаицизмом.
Причины и факторы риска
Трисомия по х хромосоме обычно не наследуется. Это происходит, когда репродуктивная клетка, имеет две Х-хромосомы из-за их нераспределения во время образования. Когда одна из таких клеток участвует в образовании зиготы, это приводит к синдрому тройного Х.
Причина мозаичной формы 46, XX / 47, XXX обусловлена аномальным делением клеток во время ранней эмбриональной стадии, что приводит к дополнительной Х-хромосоме только в некоторых клетках. Он также не является наследственным.
Симптомы и признаки
Симптомы и признаки синдрома трисомии Х широко варьируются среди пациентов. Пораженные женщины могут быть бессимптомными или иметь несколько симптомов или много аномалий. Ниже приведены аномалии, наблюдающиеся при трисомии по х хромосоме.
Узнать больше Все о синдроме титце
- Рост больше, чем средний, с длинными ногами.
- Задержка развития моторных навыков, таких как ходьба и сидение.
- Слабый мышечный тонус (гипотония).
- Низкий IQ: на 10-15 баллов ниже, чем у братьев, сестер.
- Отсроченные речевые и языковые навыки.
- Поведенческие и эмоциональные проблемы.
- Дефицит памяти, суждений, обработки информации.
- Маленькие пальцы или аномально кривые, называются клинодактильными.
- Младенцы с тройным X могут иметь эпикантальные складки (часть верхнего века, которая образует складку и покрывает внутренний угол глаза), гипертелоризм (увеличение пространства между двумя глазами), небольшая окружность головы.
- Тревожность.
- Гиперактивное расстройство с дефицитом внимания(ADHD): дети с СДВГ проявляют чрезмерную активность, отсутствие внимания, неконтролируемое поведение.
- Аномальное развитие яичников (дисвразия яичников).
- Раннее или отсроченное половое созревание.
- Преждевременная недостаточность яичников, бесплодие.
- Почечная агенезия (неспособность развиться) почечная дисплазия (аномальное развитие).
- Периодические инфекции мочевых путей.
- Плоскостопие.
- Запор, боль в животе.
- Pectus excavatum (аномальная грудная стенка, которая вогнута или утоплена)
- Сердечные аномалии.
Диагностика
Предполагается, что синдром трисомии Х возникает, когда пациент представляет любой из симптомов или задержку полового созревания или другие нарушения менструального цикла.
Хромосомный анализ
Анализ хромосом в клетках крови пострадавшего подтверждает диагноз в подозреваемых случаях.
Другие диагностические методы включают в себя пренатальную диагностику, которая выполняется у некоторых пациентов по другим причинам, а состояние диагностируется случайно.
Амниоцентез
У беременных женщин проводится проверка хромосомных аномалий растущего плода. Это инвазивная процедура. Амниотическая жидкость содержит клетки плода. Жидкость собирают и клетки исследуют, чтобы проверить количество хромосом и другие аномалии.
Если у плода есть синдром тройного Х, у клеток будет дополнительная Х-хромосома.
Выбор хорионических ворсинок
Выборка хорионических ворсинок (CVS) выполняется у беременных женщин, чтобы проверить хромосомные аномалии растущего плода. В плаценте присутствуют хорионические ворсинки. Собирают немного ткани плаценты и проверяют на аномалии хромосом. Если у плода есть трисомия по х хромосоме, у клеток будет дополнительная Х-хромосома.
Узнать больше Тетра-амелия: симптомы, признаки, лечение
Лечение
Лечение синдрома тройного Х зависит от возраста представления заболевания, его тяжести и симптомов.
Дети
Если новорожденному диагностирован синдром трисомии Х, ребенок должен оцениваться следующим образом:
- Первые 4 месяца: оценка развития мышечного тонуса и силы.
- До 12 месяцев: оценка языка, речи.
- В дошкольном возрасте: предварительная оценка ранних признаков проблем чтения.
- Для детей с синдромом тройного Х необходимо оценить функцию почек и сердца.
Детям с синдромом тройного Х, ранняя оценка и вмешательство дают отличный результат. Речевая, развивающая терапия, физиотерапия, консультирование являются ключевыми мерами вмешательства, когда это необходимо.
Молодые девушки
Для девочек с синдромом тройного Х подростковый возраст может быть сложной фазой жизни. Для них требуется короткий период консультирования.
Женщины
Для женщин с бесплодием и нарушениями менструального цикла требуется тщательная проверка наличия первичной овариальной недостаточности.
Генетическое консультирование
Генетическая консультация среди пострадавших и членов их семей является полезной.
Профилактика
Синдром трисомии X не является предупреждаемым.
Часто задаваемые вопросы
- С каким специалистом надо консультироваться, чтобы исключить синдром Triple X?
В зависимости от возраста ребенка вам может потребоваться консультация педиатра или гинекологом по проблемам, связанным с половым созреванием. Если они будут подозревать синдром Triple X, вас направят к генетику для хромосомного анализа и кариотипирования.
Является ли он наследственным расстройством?
Тройной синдром X не наследуется.
Как можно его избежать
Синдром Triple X не является предупреждаемым. Ранняя диагностика и лечение имеют решающее значение для предотвращения долгосрочных последствий.